

Scrapie-like prion protein accumulates in aggresomes of cyclosporin A-treated cells
- PMID: 12554642
- PMCID: PMC140730
- DOI: 10.1093/emboj/cdg045
Scrapie-like prion protein accumulates in aggresomes of cyclosporin A-treated cells
Abstract
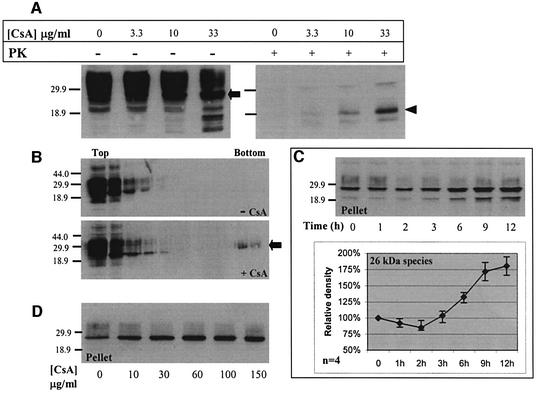
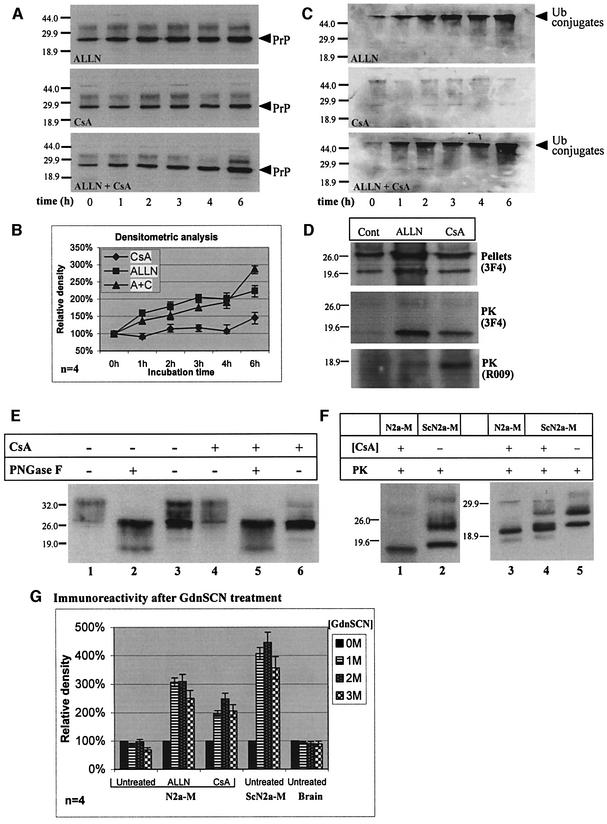
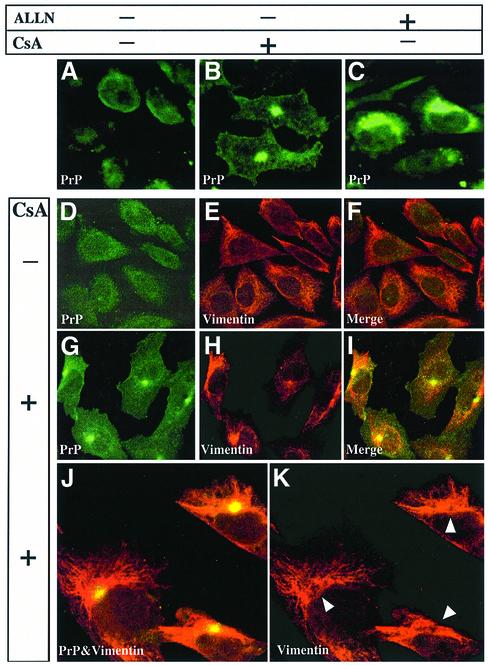
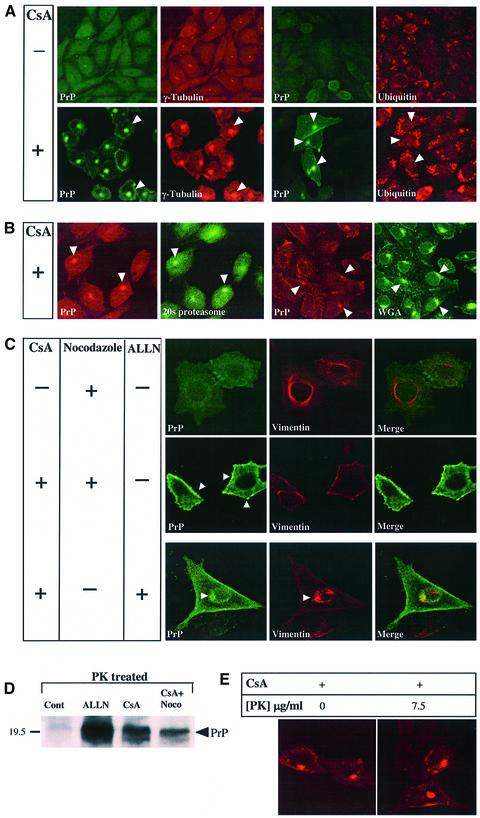
Prion diseases are infectious, sporadic and inherited fatal neurodegenerations that are propagated by an abnormal refolding of the cellular prion protein PrP(C). Which chaperones assist the normal folding of PrP(C) is unknown. The linkage of familial Gerstmann- Sträussler-Scheinker (GSS) syndrome with proline substitutions in PrP raised the prospect that peptidylprolyl cis-trans isomerases (PPIases) may play a role in normal PrP metabolism. Here we used cyclo sporin A (CsA), an immunosuppressant, to inhibit the cyclophilin family of PPIases in cultured cells. CsA-treated cells accumulated proteasome-resistant, 'prion-like' PrP species, which deposited in long-lived aggresomes. PrP aggresomes also formed with disease-linked proline mutants when proteasomes were inhibited. These results suggest mechanisms whereby abnormally folded cytosolic PrP may in some cases participate in the development of spontaneous and inherited prion diseases.
Figures
References
-
- Atkinson K., Biggs,J., Darveniza,P., Boland,J., Concannon,A. and Dodds,A. (1984) Cyclosporin-associated central nervous system toxicity after allogeneic bone marrow transplantation. Transplanta tion, 38, 34–37. - PubMed
-
- Balbach J. and Schmid,F.X. (2000) Proline isomerization and its catalysis in protein folding. In Pain,R.H (ed.), Mechanisms of Protein Folding. Oxford University Press, Oxford, UK, pp. 212–249.
-
- Basler K., Oesch,B., Scott,M., Westaway,D., Walchli,M., Groth,D.F., McKinley,M.P., Prusiner,S.B. and Weissmann,C. (1986) Scrapie and cellular PrP isoforms are encoded by the same chromosomal gene. Cell, 46, 417–428. - PubMed
-
- Bence N.F., Sampat,R.M. and Kopito,R.R. (2001) Impairment of the ubiquitin–proteasome system by protein aggregation. Science, 292, 1552–1555. - PubMed
-
- Bolton D.C., McKinley,M.P. and Prusiner,S.B. (1982) Identification of a protein that purifies with the scrapie prion. Science, 218, 1309–1311. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
