

A 100-kD complex of two RNA-binding proteins from mitochondria of Leishmania tarentolae catalyzes RNA annealing and interacts with several RNA editing components
- PMID: 12554877
- PMCID: PMC1370371
- DOI: 10.1261/rna.2134303
A 100-kD complex of two RNA-binding proteins from mitochondria of Leishmania tarentolae catalyzes RNA annealing and interacts with several RNA editing components
Abstract
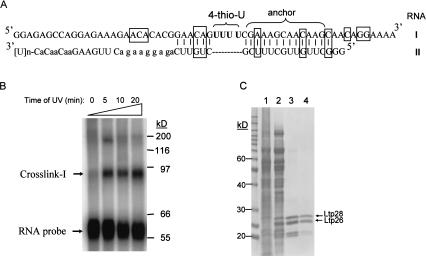
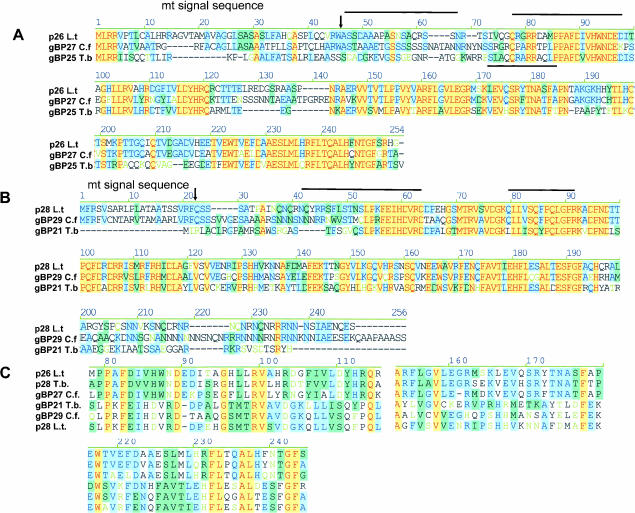
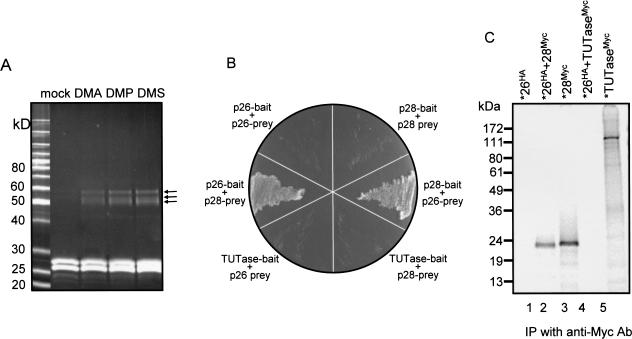
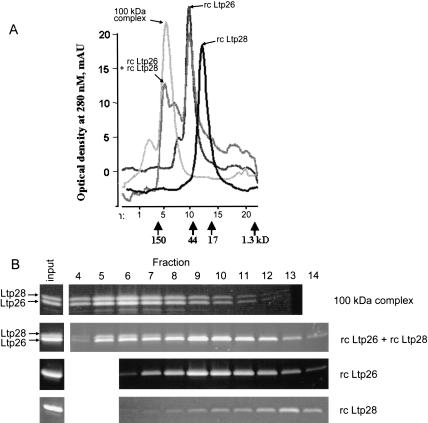
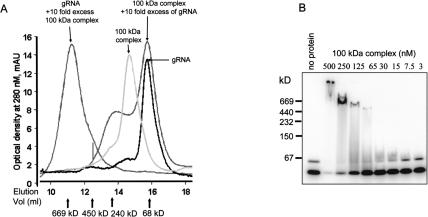
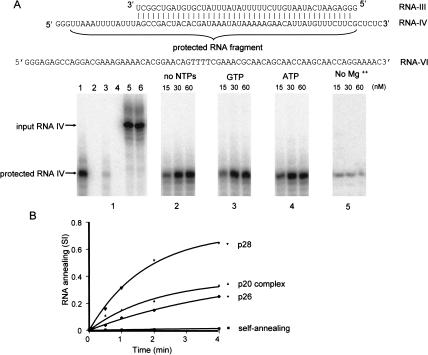
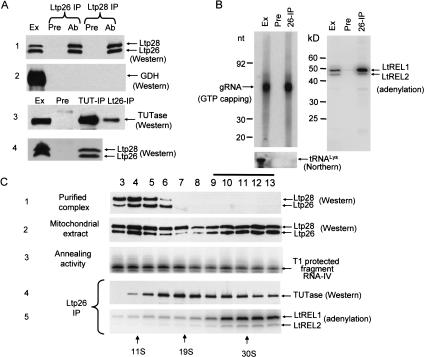
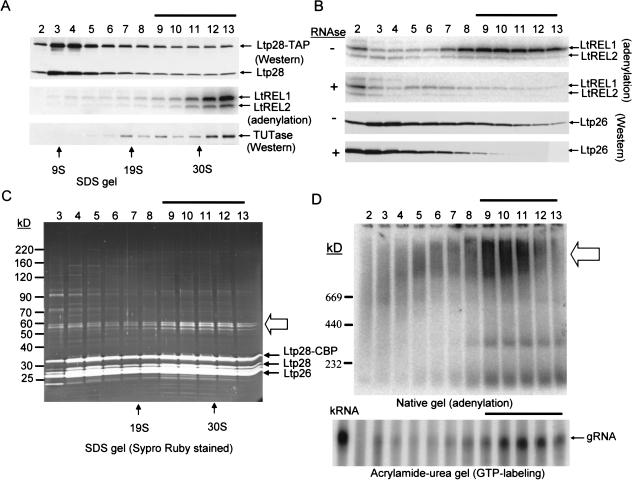
A stable 100-kD complex from mitochondria of Leishmania tarentolae containing two RNA-binding proteins, Ltp26 and Ltp28, was identified by cross-linking to unpaired 4-thiouridine nucleotides in a partially duplex RNA substrate. The genes were cloned and expressed and the complex was reconstituted from recombinant proteins in the absence of RNA or additional factors. The Ltp26 and Ltp28 proteins are homologs of gBP27 and gBP29 from Crithidia fasciculata and gBP25 and gBP21 from Trypanosoma brucei, respectively. The purified Ltp26/Ltp28 complex, the individual recombinant proteins, and the reconstituted complex are each capable of catalyzing the annealing of complementary RNAs, as was previously shown for gBP21 from T. brucei. A high-molecular-weight RNP complex consisting of the Ltp26/Ltp28 complex and several 55-60-kD proteins together with guide RNA could be purified from mitochondrial extract of L. tarentolae transfected with Ltp28-TAP. This complex also interacted in a less stable manner with the RNA ligase-containing L-complex and with the 3' TUTase. The Ltp26/Ltp28 RNP complex is a candidate for catalyzing the annealing of guide RNA and pre-edited mRNA in the initial step of RNA editing.
Figures
Similar articles
-
Isolation of a U-insertion/deletion editing complex from Leishmania tarentolae mitochondria.EMBO J. 2003 Feb 17;22(4):913-24. doi: 10.1093/emboj/cdg083. EMBO J. 2003. PMID: 12574127 Free PMC article.
-
Cloning and characterization of two guide RNA-binding proteins from mitochondria of Crithidia fasciculata: gBP27, a novel protein, and gBP29, the orthologue of Trypanosoma brucei gBP21.Nucleic Acids Res. 2001 Jul 15;29(14):2950-62. doi: 10.1093/nar/29.14.2950. Nucleic Acids Res. 2001. PMID: 11452020 Free PMC article.
-
Reconstitution of full-round uridine-deletion RNA editing with three recombinant proteins.Proc Natl Acad Sci U S A. 2006 Sep 19;103(38):13944-9. doi: 10.1073/pnas.0604476103. Epub 2006 Sep 8. Proc Natl Acad Sci U S A. 2006. PMID: 16963561 Free PMC article.
-
RNA editing in mitochondria of Leishmania tarentolae and Crithidia fasciculata.Semin Cell Biol. 1993 Aug;4(4):241-9. doi: 10.1006/scel.1993.1029. Semin Cell Biol. 1993. PMID: 7694672 Review.
-
RNA editing in kinetoplastid mitochondria.FASEB J. 1993 Jan;7(1):54-63. doi: 10.1096/fasebj.7.1.8422975. FASEB J. 1993. PMID: 8422975 Review.
Cited by
-
Interactions of mRNAs and gRNAs involved in trypanosome mitochondrial RNA editing: structure probing of a gRNA bound to its cognate mRNA.RNA. 2006 Jun;12(6):1050-60. doi: 10.1261/rna.3406. Epub 2006 Apr 17. RNA. 2006. PMID: 16618968 Free PMC article.
-
Isolation of a U-insertion/deletion editing complex from Leishmania tarentolae mitochondria.EMBO J. 2003 Feb 17;22(4):913-24. doi: 10.1093/emboj/cdg083. EMBO J. 2003. PMID: 12574127 Free PMC article.
-
Uridine insertion/deletion RNA editing in trypanosome mitochondria: a complex business.RNA. 2003 Mar;9(3):265-76. doi: 10.1261/rna.2178403. RNA. 2003. PMID: 12591999 Free PMC article. Review.
-
Lexis and Grammar of Mitochondrial RNA Processing in Trypanosomes.Trends Parasitol. 2020 Apr;36(4):337-355. doi: 10.1016/j.pt.2020.01.006. Epub 2020 Feb 28. Trends Parasitol. 2020. PMID: 32191849 Free PMC article. Review.
-
Transcription initiation defines kinetoplast RNA boundaries.Proc Natl Acad Sci U S A. 2018 Oct 30;115(44):E10323-E10332. doi: 10.1073/pnas.1808981115. Epub 2018 Oct 17. Proc Natl Acad Sci U S A. 2018. PMID: 30333188 Free PMC article.
References
-
- Aphasizhev, R. and Simpson, L. 2001. Isolation and characterization of a U-specific 3′-5′ exonuclease from mitochondria of Leishmania tarentolae. J. Biol. Chem. 276: 21280–21284. - PubMed
-
- Aphasizhev, R., Sbicego, S., Peris, M., Jang, S.H., Aphasizheva, I., Simpson, A.M., Rivlin, A., and Simpson, L. 2002. Trypanosome mitochondrial 3′ terminal uridylyl transferase (TUTase): The key enzyme in U-insertion/deletion RNA editing. Cell 108: 637–648. - PubMed
-
- Blom, D., Burg, J., Breek, C.K., Speijer, D., Muijsers, A.O., and Benne, R. 2001. Cloning and characterization of two guide RNA-binding proteins from mitochondria of Crithidia fasciculata: gBP27, a novel protein, and gBP29, the orthologue of Trypanosoma brucei gBP21. Nucleic Acids Res. 29: 2950–2962. - PMC - PubMed
-
- Blum, B., Bakalara, N., and Simpson, L. 1990. A model for RNA editing in kinetoplastid mitochondria: “Guide” RNA molecules transcribed from maxicircle DNA provide the edited information. Cell 60: 189–198. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous