

Rho family GTPases are required for activation of Jak/STAT signaling by G protein-coupled receptors
- PMID: 12556491
- PMCID: PMC141129
- DOI: 10.1128/MCB.23.4.1316-1333.2003
Rho family GTPases are required for activation of Jak/STAT signaling by G protein-coupled receptors
Abstract
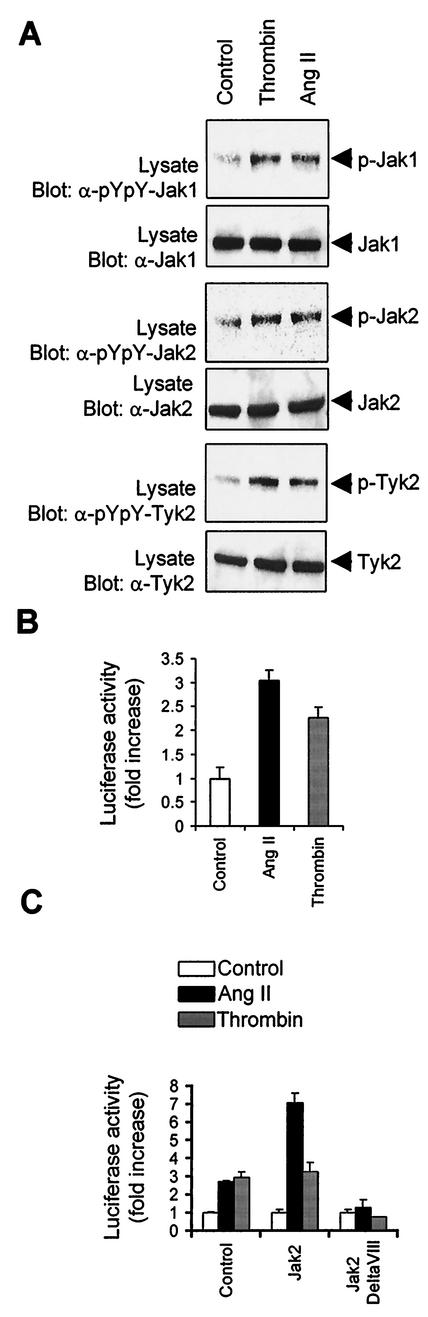
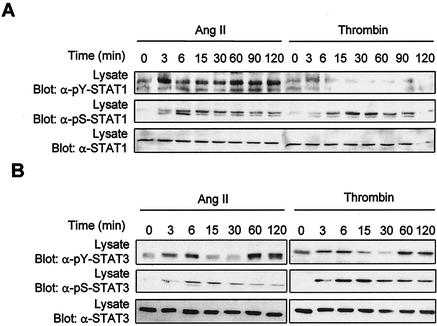
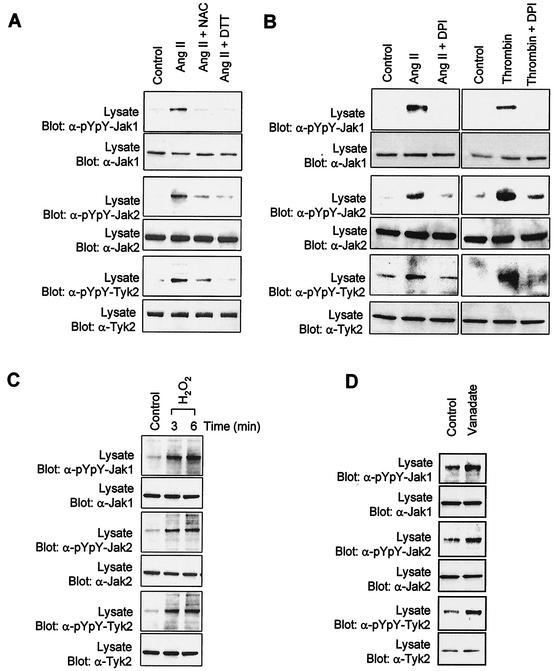
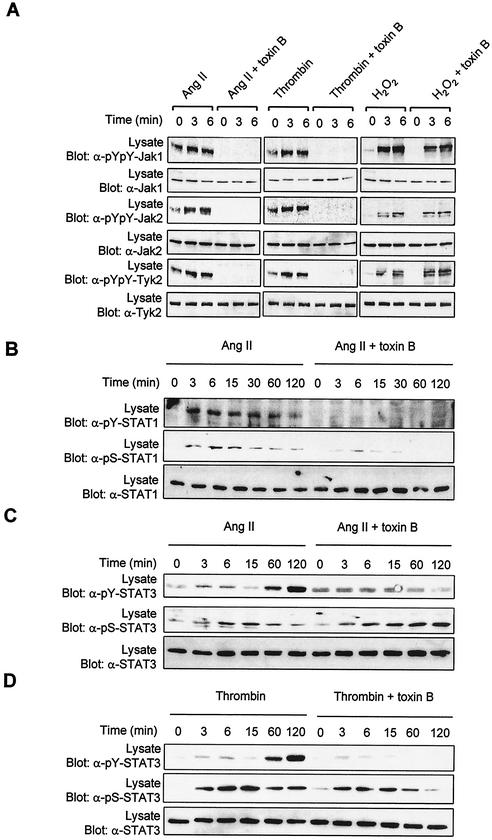
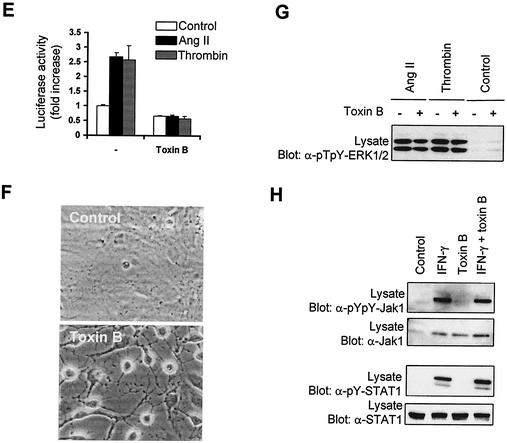
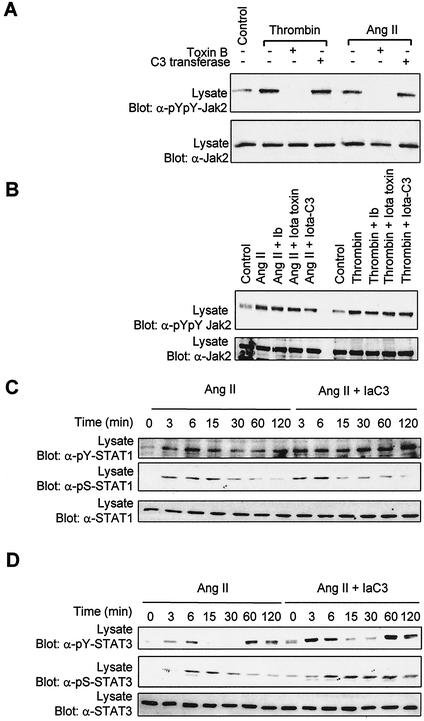
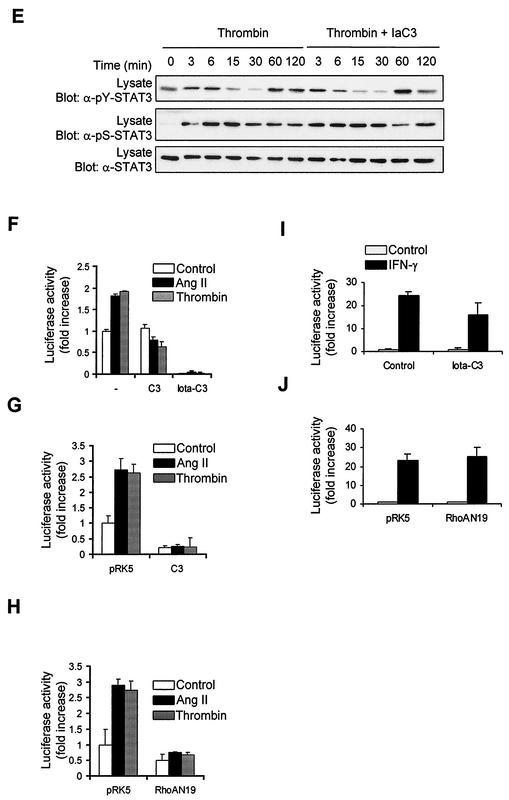
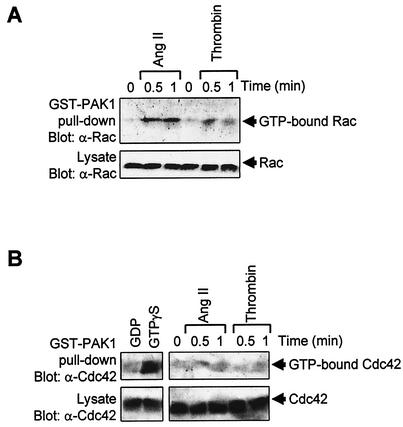
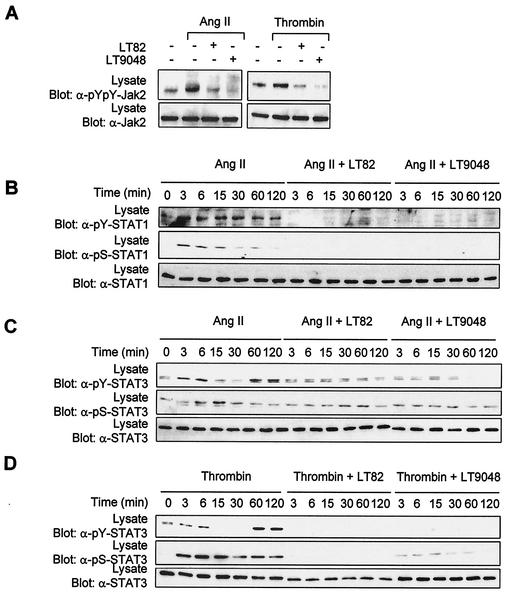
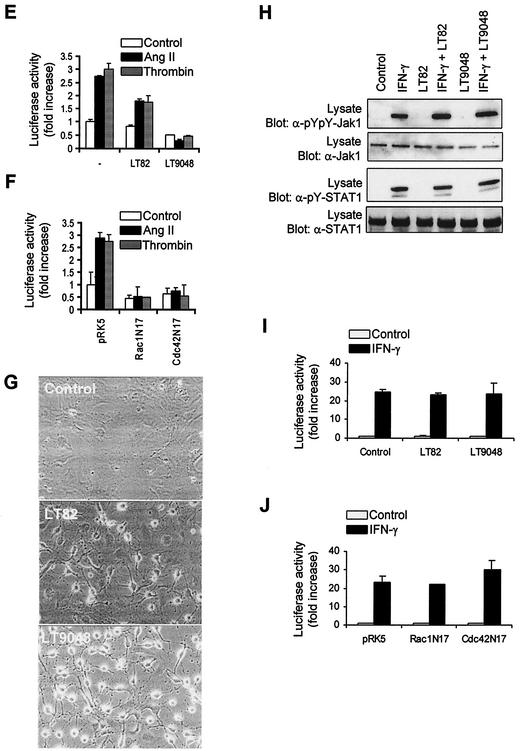
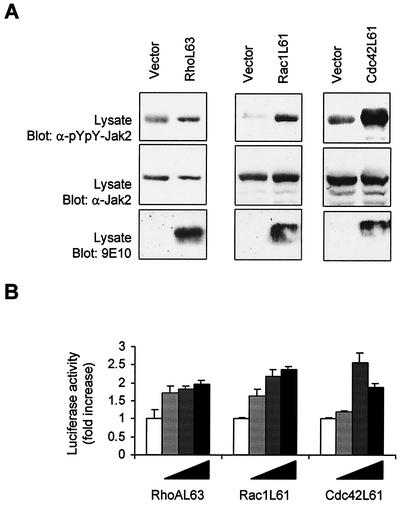
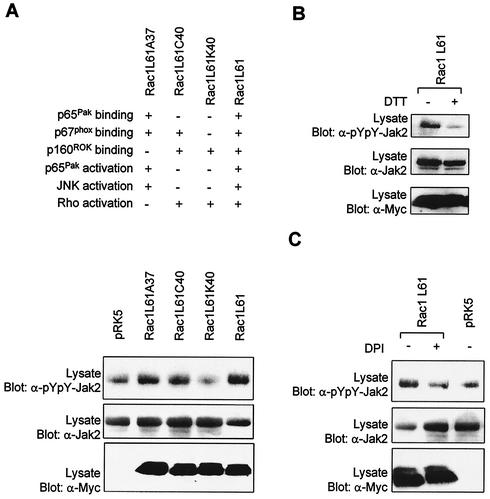
As do cytokine receptors and receptor tyrosine kinases, G protein-coupled receptors (GPCRs) signal to Janus kinases (Jaks) and signal transducers and activators of transcription (STATs). However, the early biochemical events linking GPCRs to this signaling pathway have been unclear. Here we show that GPCR-stimulated Rac activity and the subsequent generation of reactive oxygen species are necessary for activating tyrosine phosphorylation of Jaks and STAT-dependent transcription. The requirement for Rac activity can be overcome by addition of hydrogen peroxide. Expression of activated mutants of Rac1 is sufficient to activate Jak2 and STAT-dependent transcription, and the activation of Jak2 correlates with the ability of Rac1 to bind to NADPH oxidase subunit p67(phox). We further show that GPCR agonists stimulate tyrosine phosphorylation of STAT1 and STAT3 proteins in a Rac-dependent manner. The tyrosine phosphorylation of STAT3 is biphasic; the first peak of phosphorylation is weak and correlates with rapid activation of Jaks by GPCRs, whereas the second peak is stronger and requires the synthesis of an autocrine factor. Rho also plays an essential role in the induction of STAT transcriptional activity. Our results highlight a novel role for Rho GTPases in mediating the regulatory effects of GPCRs on STAT-dependent gene expression.
Figures
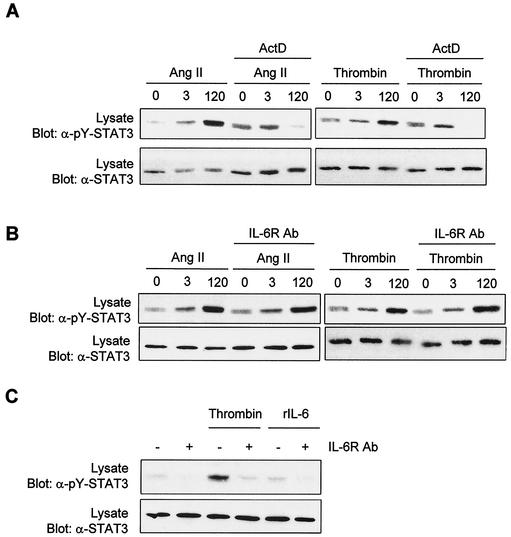
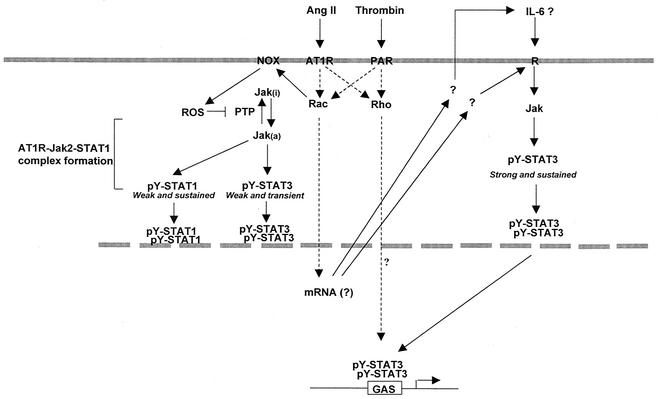
References
-
- Aktories, K., G. Schmidt, and I. Just. 2000. Rho GTPases as targets of bacterial protein toxins. Biol. Chem. 381:421-426. - PubMed
-
- Ali, M. S., P. P. Sayeski, and K. E. Bernstein. 2000. Jak2 acts as both a STAT1 kinase and as a molecular bridge linking STAT1 to the angiotensin II AT1 receptor. J. Biol. Chem. 275:15586-15593. - PubMed
-
- Ali, M. S., P. P. Sayeski, L. B. Dirksen, D. J. Hayzer, M. B. Marrero, and K. E. Bernstein. 1997. Dependence on the motif YIPP for the physical association of Jak2 kinase with the intracellular carboxyl tail of the angiotensin II AT1 receptor. J. Biol. Chem. 272:23382-23388. - PubMed
-
- Ali, M. S., P. P. Sayeski, A. Safavi, M. Lyles, and K. E. Bernstein. 1998. Janus kinase 2 (Jak2) must be catalytically active to associate with the AT1 receptor in response to angiotensin II. Biochem. Biophys. Res. Commun. 249:672-677. - PubMed
-
- Babior, B. M. 1999. NADPH oxidase: an update. Blood 93:1464-1476. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous