

Human population genetic structure and inference of group membership
- PMID: 12557124
- PMCID: PMC1180234
- DOI: 10.1086/368061
Human population genetic structure and inference of group membership
Abstract
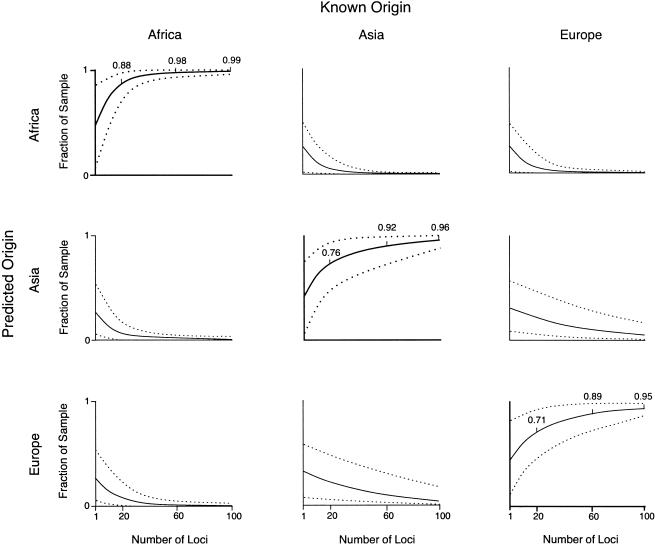
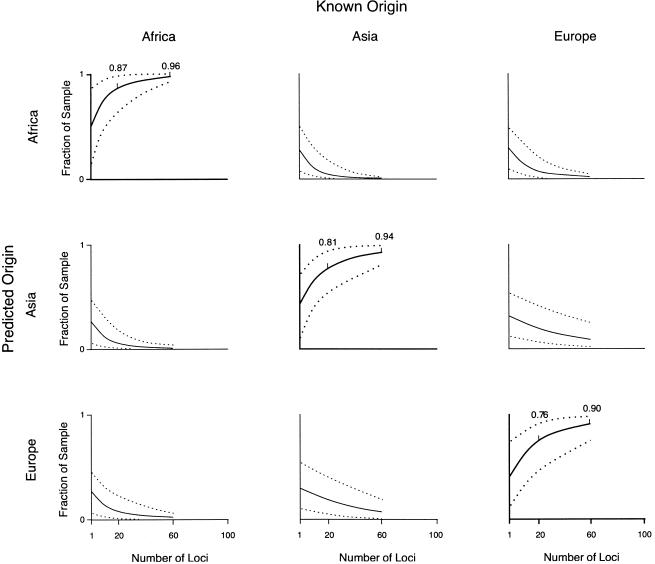
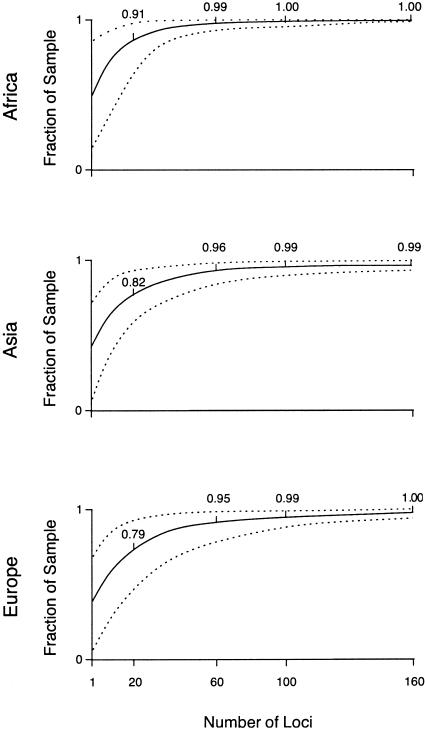
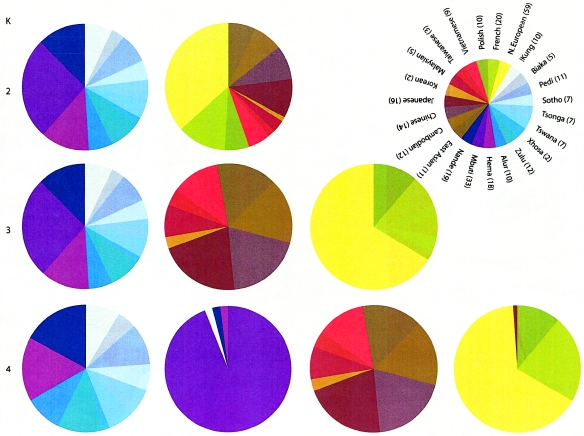
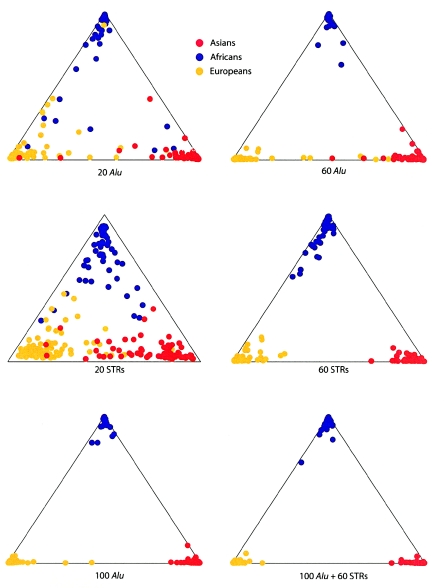
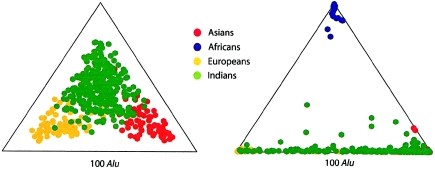
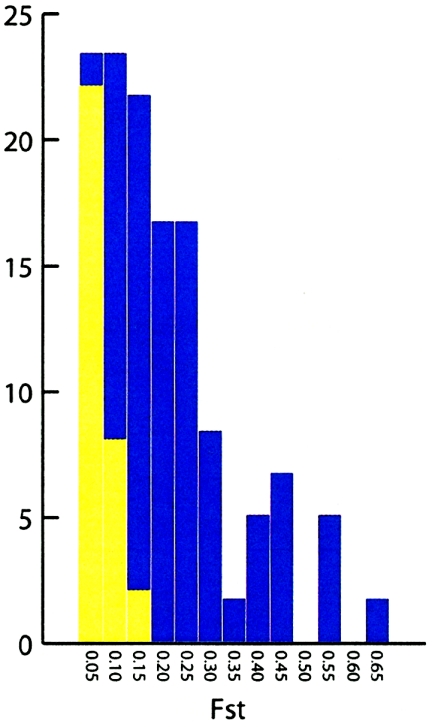
A major goal of biomedical research is to develop the capability to provide highly personalized health care. To do so, it is necessary to understand the distribution of interindividual genetic variation at loci underlying physical characteristics, disease susceptibility, and response to treatment. Variation at these loci commonly exhibits geographic structuring and may contribute to phenotypic differences between groups. Thus, in some situations, it may be important to consider these groups separately. Membership in these groups is commonly inferred by use of a proxy such as place-of-origin or ethnic affiliation. These inferences are frequently weakened, however, by use of surrogates, such as skin color, for these proxies, the distribution of which bears little resemblance to the distribution of neutral genetic variation. Consequently, it has become increasingly controversial whether proxies are sufficient and accurate representations of groups inferred from neutral genetic variation. This raises three questions: how many data are required to identify population structure at a meaningful level of resolution, to what level can population structure be resolved, and do some proxies represent population structure accurately? We assayed 100 Alu insertion polymorphisms in a heterogeneous collection of approximately 565 individuals, approximately 200 of whom were also typed for 60 microsatellites. Stripped of identifying information, correct assignment to the continent of origin (Africa, Asia, or Europe) with a mean accuracy of at least 90% required a minimum of 60 Alu markers or microsatellites and reached 99%-100% when >/=100 loci were used. Less accurate assignment (87%) to the appropriate genetic cluster was possible for a historically admixed sample from southern India. These results set a minimum for the number of markers that must be tested to make strong inferences about detecting population structure among Old World populations under ideal experimental conditions. We note that, whereas some proxies correspond crudely, if at all, to population structure, the heuristic value of others is much higher. This suggests that a more flexible framework is needed for making inferences about population structure and the utility of proxies.
Figures
References
Electronic-Database Information
-
- Lewis Lab Software, http://lewis.eeb.uconn.edu/lewishome/software.html (for Genetic Data Analysis [GDA], the software used to estimate FST statistics)
-
- M.A.B.'s Web site, http://batzerlab.lsu.edu/ (for the specific identities of each Alu marker, the PCR conditions used to amplify each system, and the expected amplicon sizes)
-
- Pritchard Lab, http://pritch.bsd.uchicago.edu/ (for Structure, the software used to detect population structure and make inferences about population assignment)
-
- W.S.W.'s Web site, http://www.genetics.utah.edu/~swatkins/pub/Alu_data.htm (for the specific identities of each Alu marker, the PCR conditions used to amplify each system, and the expected amplicon sizes)
References
-
- Aspinall PJ (1998) Describing the “white” ethnic group and its composition in medical research. Soc Sci Med 47:1797–1808 - PubMed
-
- Bamshad M, Kivisild T, Watkins WS, Dixon ME, Ricker CE, Rao BB, Naidu JM, Prasad BV, Reddy PG, Rasanayagam A, Papiha SS, Villems R, Redd AJ, Hammer MF, Nguyen SV, Carroll ML, Batzer MA, Jorde LB (2001) Genetic evidence on the origins of Indian caste populations. Genome Res 11:994–1004 - PMC - PubMed
-
- Batzer MA, Deininger PL (2002) Alu repeats and human genomic diversity. Nat Rev Genet 3:370–379 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
