

Regulation of lymphocyte apoptosis by interferon regulatory factor 4 (IRF-4)
- PMID: 12566414
- PMCID: PMC2193834
- DOI: 10.1084/jem.20020717
Regulation of lymphocyte apoptosis by interferon regulatory factor 4 (IRF-4)
Abstract
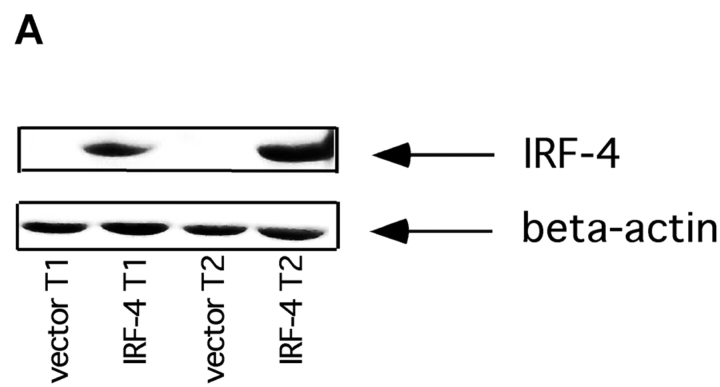
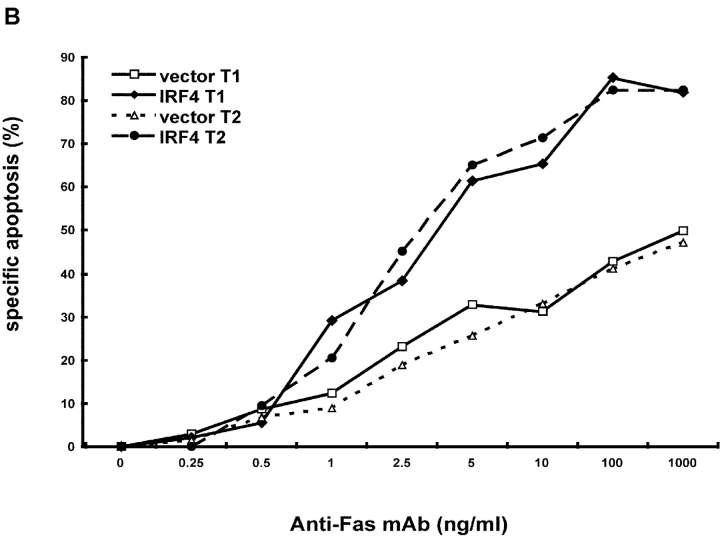
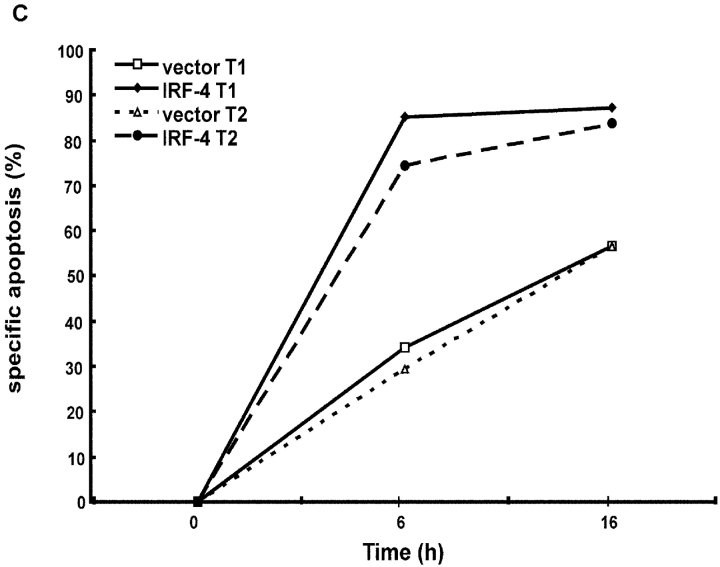
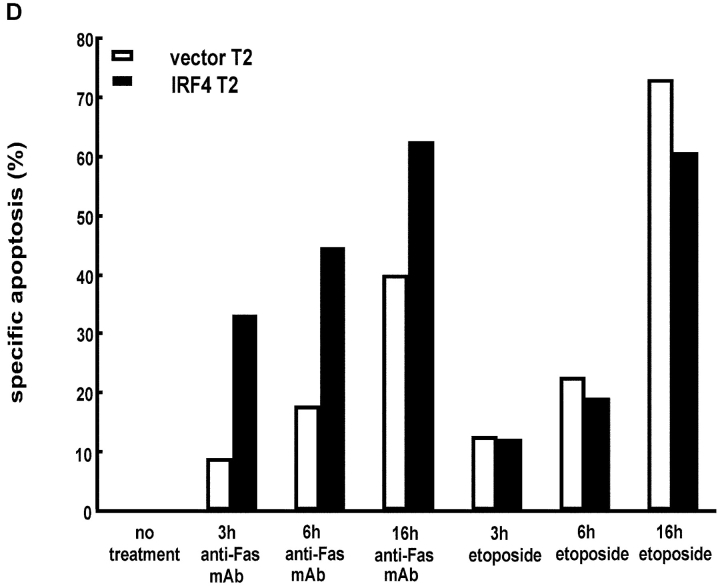
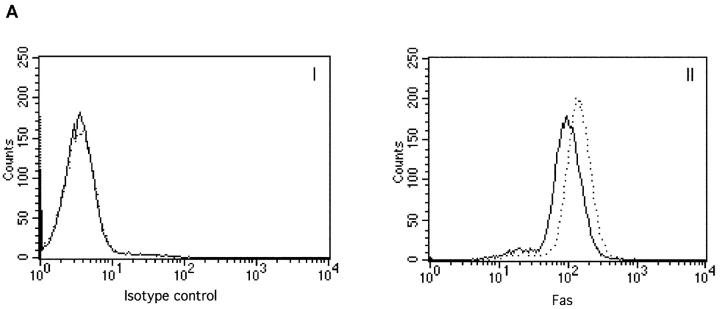
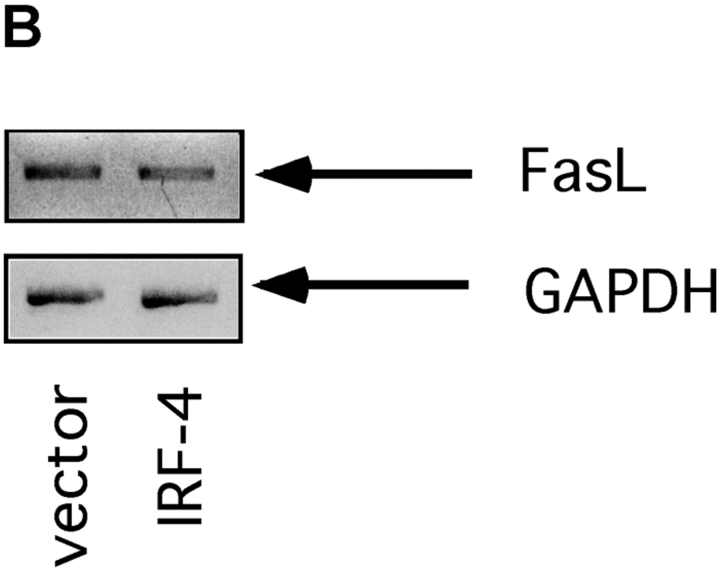
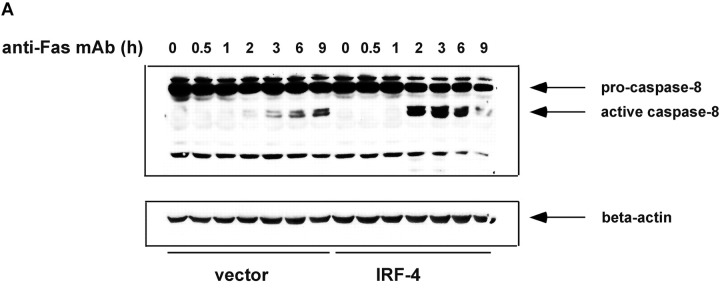
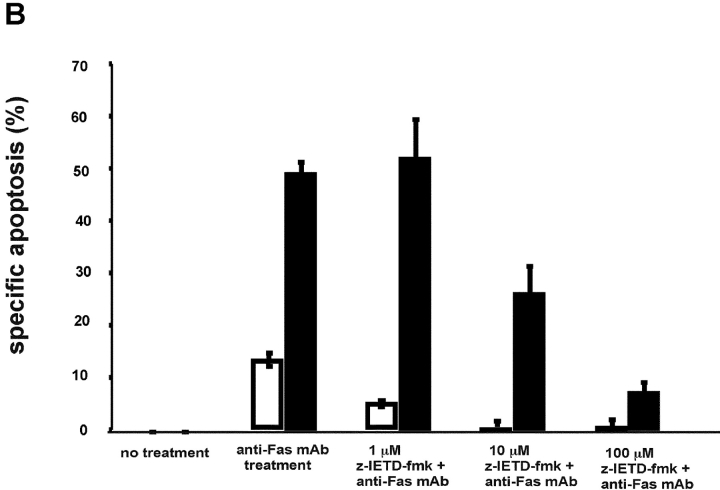
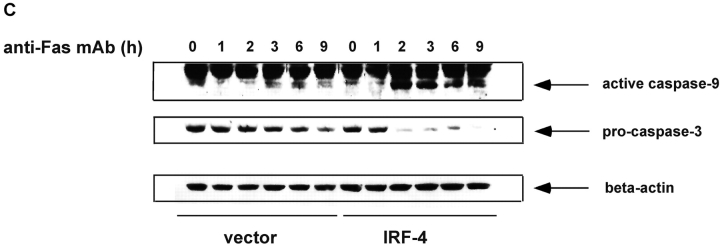
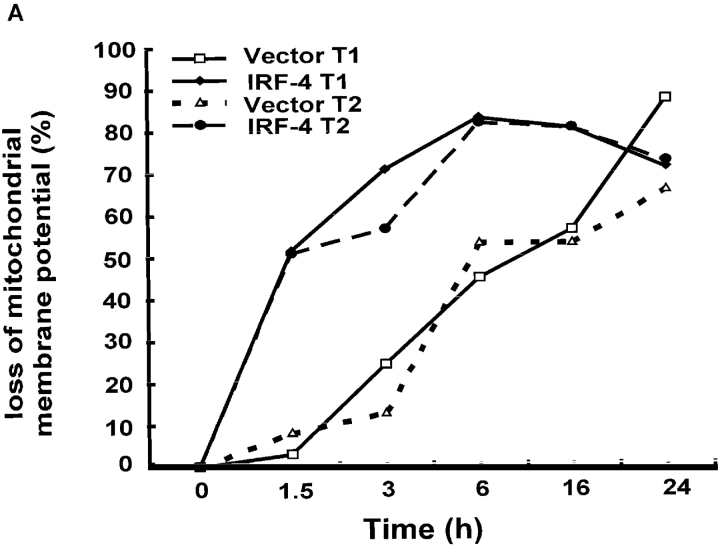
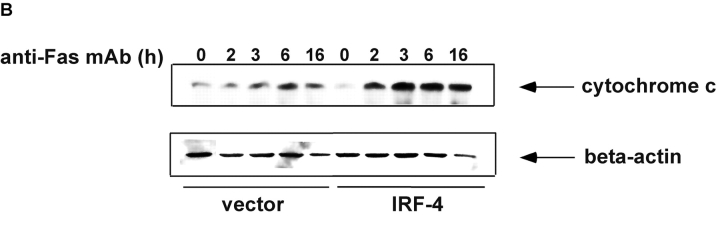
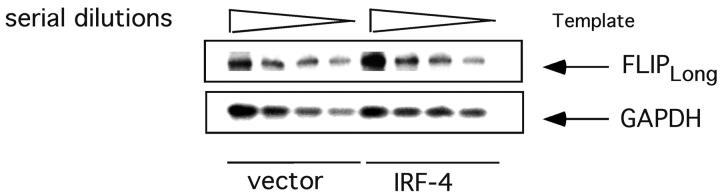
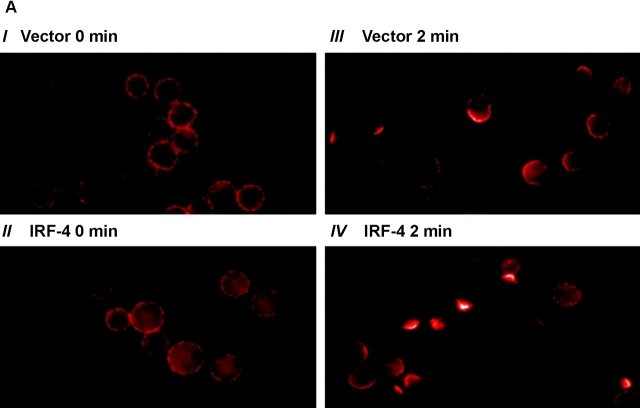
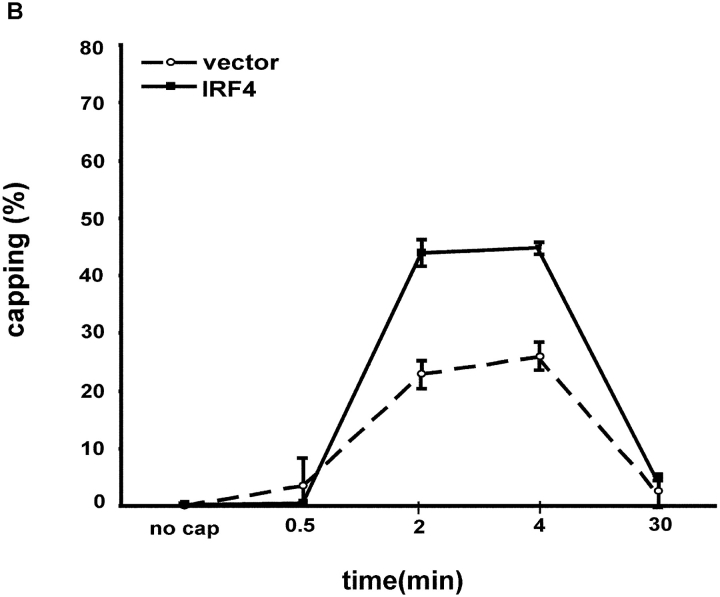
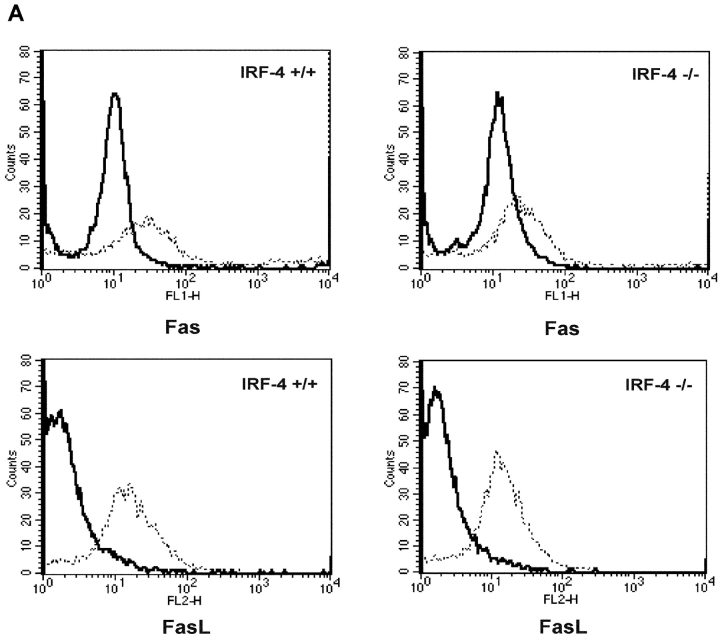
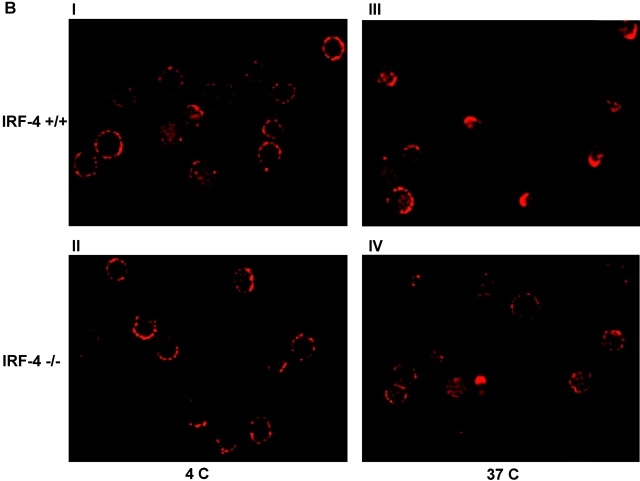
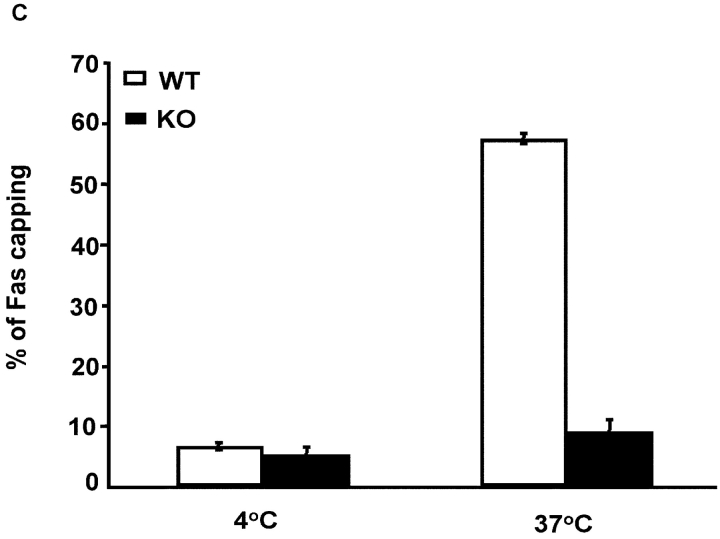
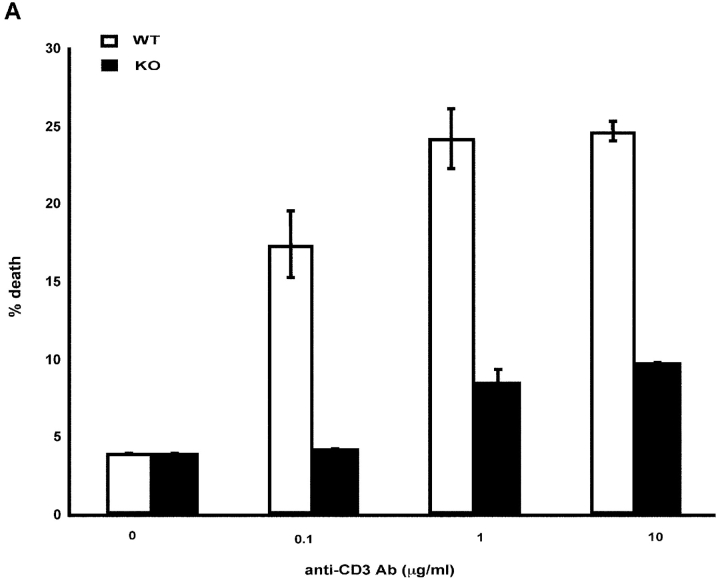
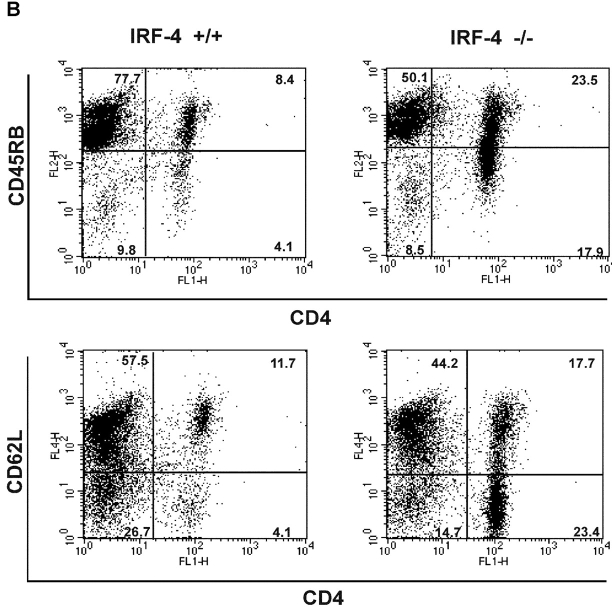
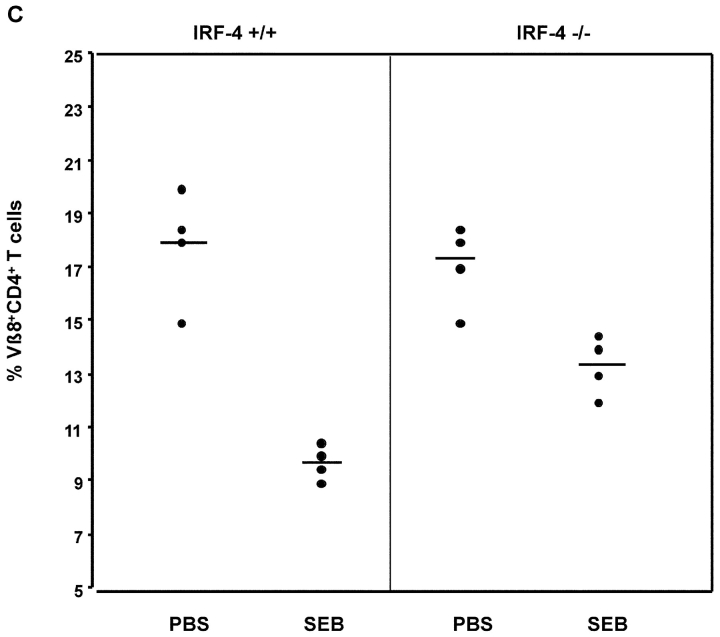
To ensure that homeostasis of the immune system is maintained, the sensitivity of lymphocytes to Fas-mediated apoptosis is differentially regulated during their activation. The molecular mechanisms that link the activation program of lymphocytes to changes in sensitivity to Fas-mediated apoptosis have, however, not been fully characterized. In these studies, we have investigated whether Fas-mediated apoptosis can be regulated by interferon regulatory factor 4 (IRF-4), a lymphoid-restricted member of the IRF family of transcription factors. IRF-4 expression is upregulated during lymphocyte activation and IRF-4-deficient mice have defects in both lymphocyte activation and homeostasis. Here, we show that stable expression of IRF-4 in a human lymphoid cell line that normally lacks IRF-4 leads to a significantly enhanced apoptotic response on Fas receptor engagement. A systematic examination of the downstream effectors of Fas signaling in IRF-4-transfected cells demonstrates an increased activation of caspase-8, as well as an increase in Fas receptor polarization. We demonstrate that IRF-4-deficient mice display defects in activation-induced cell death, as well as superantigen-induced deletion, and that these defects are accompanied by impairments in Fas receptor polarization. These data suggest that IRF-4, by modulating the efficiency of the Fas-mediated death signal, is a novel participant in the regulation of lymphoid cell apoptosis.
Figures
References
-
- Thome, M., and J. Tschopp. 2001. Regulation of lymphocyte proliferation and death by FLIP. Nat. Rev. Immunol. 1:50–58. - PubMed
-
- Nagata, S., and P. Golstein. 1995. The Fas death factor. Science. 267:1449–1456. - PubMed
-
- Schulze-Osthoff, K., D. Ferrari, M. Los, S. Wesselborg, and M.E. Peter. 1998. Apoptosis signaling by death receptors. Eur. J. Biochem. 254:439–459. - PubMed
-
- Golstein, P. 2000. Signal transduction. FasL binds preassembled Fas. Science. 288:2328–2329. - PubMed
-
- Muzio, M., A.M. Chinnaiyan, F.C. Kischkel, K. O'Rourke, A. Shevchenko, J. Ni, C. Scaffidi, J.D. Bretz, M. Zhang, R. Gentz, et al. 1996. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell. 85:817–827. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
