

Review
doi: 10.1172/JCI17741.
Neuronal degeneration and mitochondrial dysfunction
Affiliations
- PMID: 12569152
- PMCID: PMC151870
- DOI: 10.1172/JCI17741
Item in Clipboard
Review
Neuronal degeneration and mitochondrial dysfunction
J Clin Invest.
2003 Feb.
No abstract available
Figures
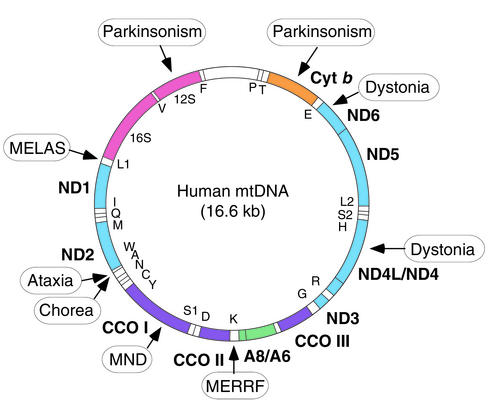
Map of the human mitochondrial genome (1). Polypeptide-coding genes (boldface) are outside the circle and specify seven subunits of NADH dehydrogenase–coenzyme Q oxidoreductase (ND), one subunit of coenzyme Q–cytochrome c oxidoreductase (Cyt b), three subunits of cytochrome c oxidase (CCO), and two subunits of ATP synthase (A) (see also Figure 2). Protein synthesis genes (12S and 16S rRNAs, and 22 tRNAs [one-letter code]) are inside the circle. Mutations in mtDNA associated with MELAS and MERRF, and mutations with features of neurodegenerative disorders, such as ataxia, chorea, dystonia, motor neuron disease (MND), and parkinsonism, are boxed. A complete list of pathogenic mtDNA mutations may be found in ref. .
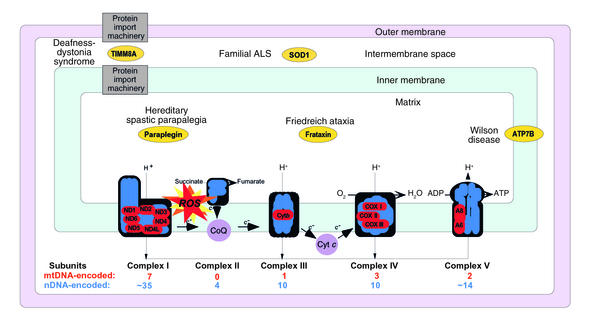
Schematic representation of the mitochondrion with its electron transport chain (ETC). The ETC is the principal source of ROS in the cell. In addition to mutations in mtDNA- and nDNA-encoded components of the ETC, a number of mutant mitochondrial proteins that do not belong to the ETC have been associated with neurodegenerative disorders. The intramitochondrial localization and the clinical phenotypes associated with mutations of some of these proteins (in ovals) are indicated.
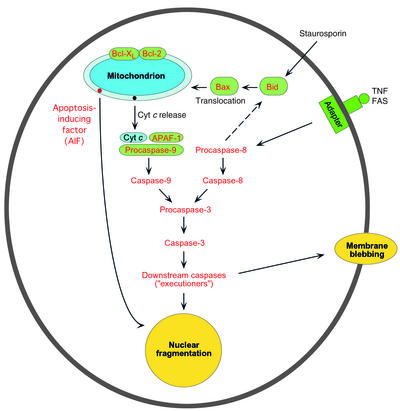
A schematic view of the main pathways of apoptosis: mitochondrion-mediated and mitochondrion-independent. See text for details.
References
-
- Anderson S, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. - PubMed
-
- Schon EA, Bonilla E, DiMauro S. Mitochondrial DNA mutations and pathogenesis. J. Bioenerg. Biomembr. 1997;29:131–149. - PubMed
-
- Cavadini P, O’Neill HA, Benada O, Isaya G. Assembly and iron-binding properties of human frataxin, the protein deficient in Friedreich ataxia. Hum. Mol. Genet. 2002;11:217–227. - PubMed
-
- Gakh O, et al. Physical evidence that yeast frataxin is an iron storage protein. Biochemistry. 2002;41:6798–6804. - PubMed
-
- Puccio H. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat. Genet. 2001;27:181–186. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
