

Identification of an endogenous ligand that activates pregnane X receptor-mediated sterol clearance
- PMID: 12569201
- PMCID: PMC298687
- DOI: 10.1073/pnas.0336235100
Identification of an endogenous ligand that activates pregnane X receptor-mediated sterol clearance
Abstract
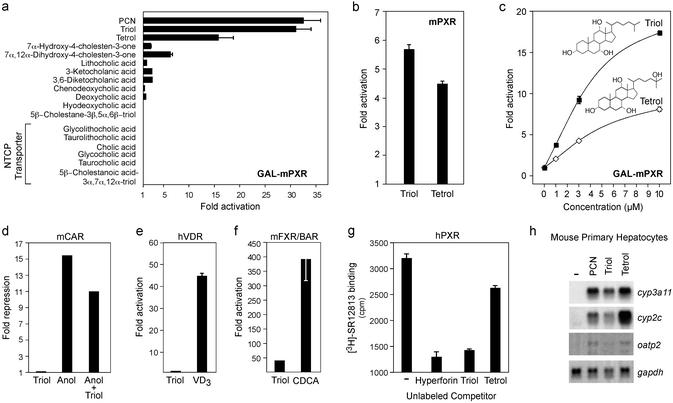
The nuclear receptor PXR (pregnane X receptor) is a broad-specificity sensor that recognizes a wide variety of synthetic drugs and xenobiotic agents. On activation by these compounds, PXR coordinately induces a network of transporters, cytochrome P450 enzymes, and other genes that effectively clear xenobiotics from the liver and intestine. Like PXR, the majority of its target genes also possess a broad specificity for exogenous compounds. Thus, PXR is both a sensor and effector in a well integrated and generalized pathway for chemical immunity. Although it is clear that PXR responds to numerous foreign compounds, it is unclear whether it possesses an endogenous ligand. To address this issue, we noted that there is substantial overlap in the substrate specificities of PXR and its critical CYP3A target gene. This prompted us to ask whether endogenous CYP3A substrates also serve as PXR ligands. We demonstrate that 5beta-cholestane-3alpha,7alpha,12alpha-triol (triol), a cholesterol-derived CYP3A substrate, is a potent PXR agonist that effectively induces cyp3a expression in mice. This defines a critical salvage pathway that can be autoinduced to minimize triol accumulation. In contrast, triol can accumulate to very high levels in humans, and unlike mice, these people develop the severe clinical manifestations of cerebrotendinous xanthomatosis. The reason for these dramatic species differences has remained unclear. We now demonstrate that triol fails to activate human PXR or induce the CYP3A-salvage pathway. This explains why humans are more susceptible to sterol accumulation and suggests that synthetic ligands for human PXR could be used to treat cerebrotendinous xanthomatosis and other disorders of cholesterol excess.
Figures


References
-
- Dussault I, Forman B M. Crit Rev Eukaryotic Gene Expression. 2002;12:53–64. - PubMed
-
- Xie W, Evans R M. J Biol Chem. 2001;276:37739–37742. - PubMed
-
- Synold T W, Dussault I, Forman B M. Nat Med. 2001;7:584–590. - PubMed
-
- Maglich J M, Stoltz C M, Goodwin B, Hawkins-Brown D, Moore J T, Kliewer S A. Mol Pharmacol. 2002;62:638–646. - PubMed
-
- Rae J M, Johnson M D, Lippman M E, Flockhart D A. J Pharmacol Exp Ther. 2001;299:849–857. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
