

Isolation of a U-insertion/deletion editing complex from Leishmania tarentolae mitochondria
- PMID: 12574127
- PMCID: PMC145443
- DOI: 10.1093/emboj/cdg083
Isolation of a U-insertion/deletion editing complex from Leishmania tarentolae mitochondria
Abstract
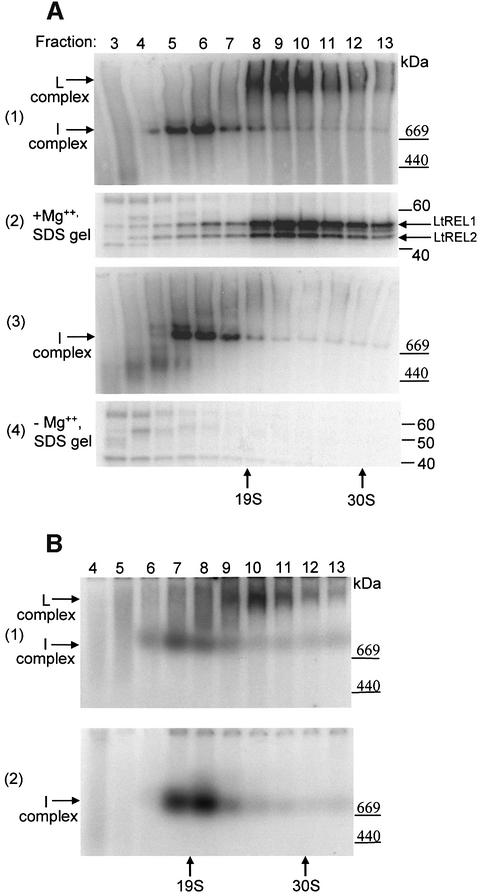
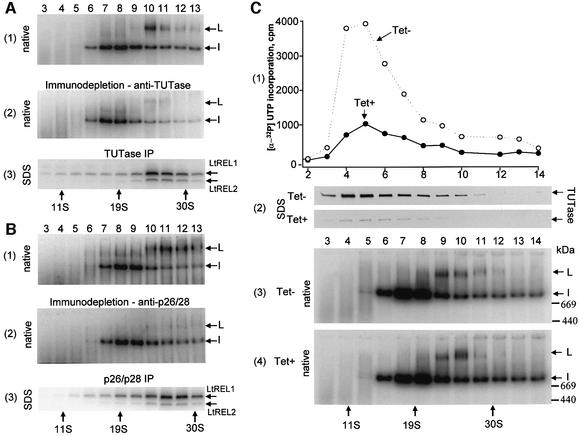
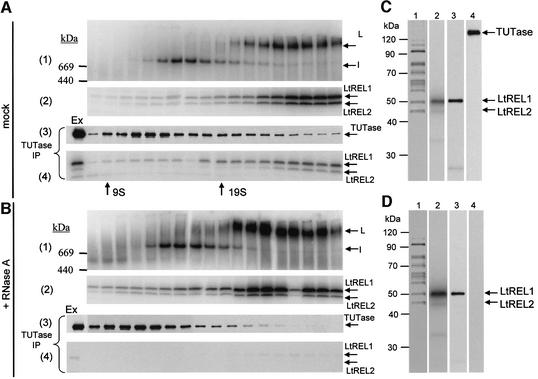
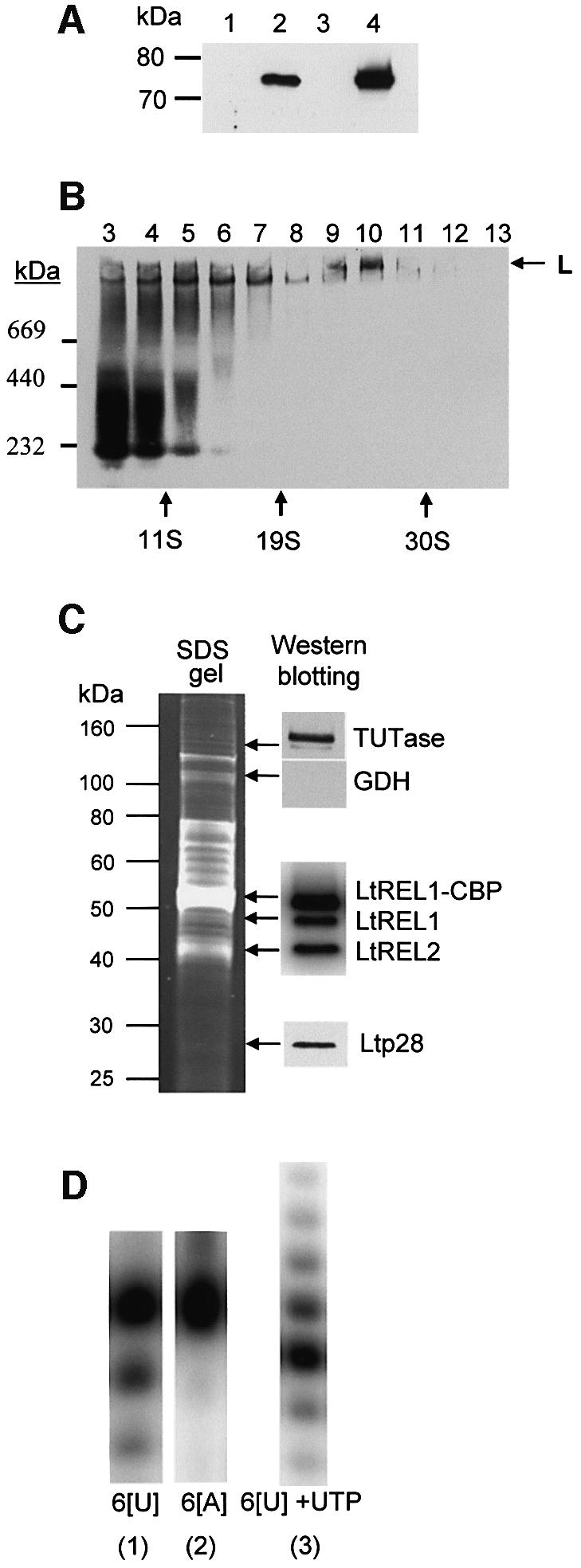
A multiprotein, high molecular weight complex active in both U-insertion and U-deletion as judged by a pre-cleaved RNA editing assay was isolated from mitochondrial extracts of Leishmania tarentolae by the tandem affinity purification (TAP) procedure, using three different TAP-tagged proteins of the complex. This editing- or E-complex consists of at least three protein-containing components interacting via RNA: the RNA ligase-containing L-complex, a 3' TUTase (terminal uridylyltransferase) and two RNA-binding proteins, Ltp26 and Ltp28. Thirteen approximately stoichiometric components were identified by mass spectrometric analysis of the core L-complex: two RNA ligases; homologs of the four Trypanosoma brucei editing proteins; and seven novel polypeptides, among which were two with RNase III, one with an AP endo/exonuclease and one with nucleotidyltransferase motifs. Three proteins have no similarities beyond kinetoplastids.
Figures
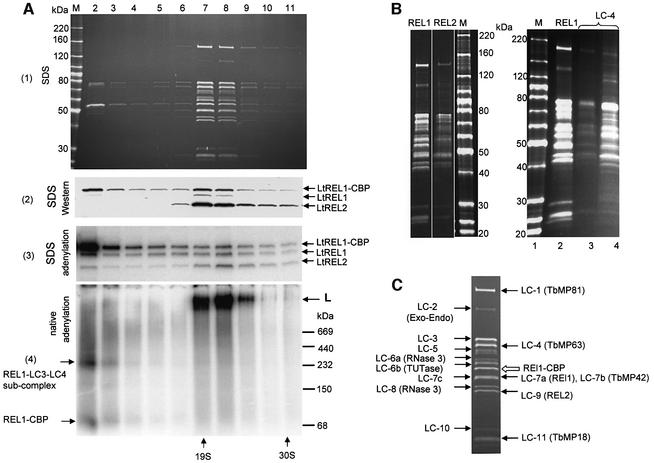
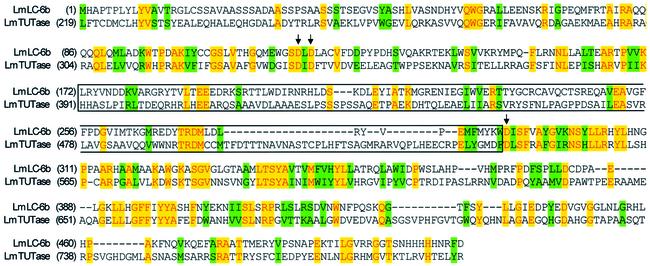
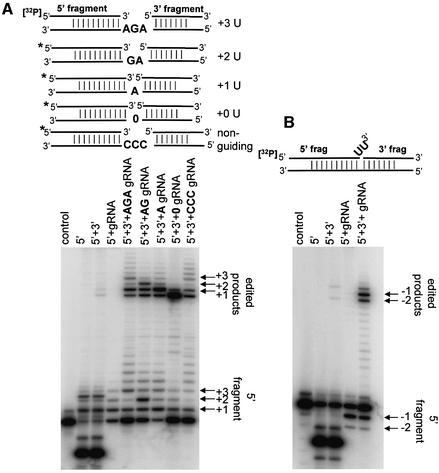
References
-
- Aphasizhev R. and Simpson,L. (2001) Isolation and characterization of a U-specific 3′–5′ exonuclease from mitochondria of Leishmania tarentolae. J. Biol. Chem., 276, 21280–21284. - PubMed
-
- Aphasizhev R., Sbicego,S., Peris,M., Jang,S.H., Aphasizheva,I., Simpson,A.M., Rivlin,A. and Simpson,L. (2002) Trypanosome mitochondrial 3′ terminal uridylyl transferase (TUTase): the key enzyme in U-insertion/deletion RNA editing. Cell, 108, 637–648. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
