

Store-operated Ca2+ entry: dynamic interplay between endoplasmic reticulum, mitochondria and plasma membrane
- PMID: 12576497
- PMCID: PMC2342659
- DOI: 10.1113/jphysiol.2002.034140
Store-operated Ca2+ entry: dynamic interplay between endoplasmic reticulum, mitochondria and plasma membrane
Abstract
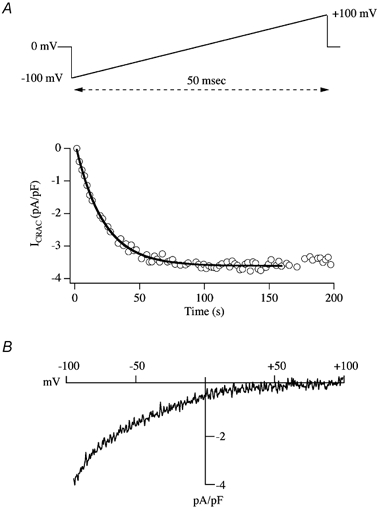
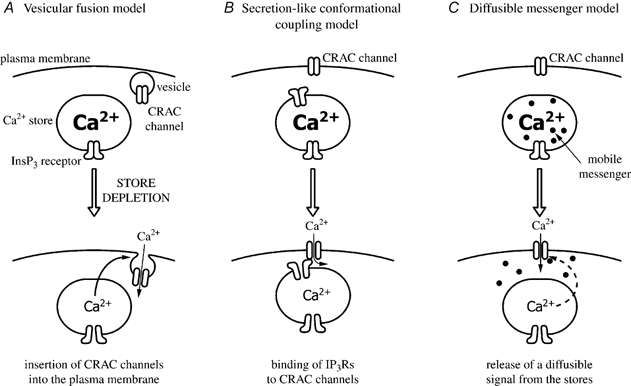
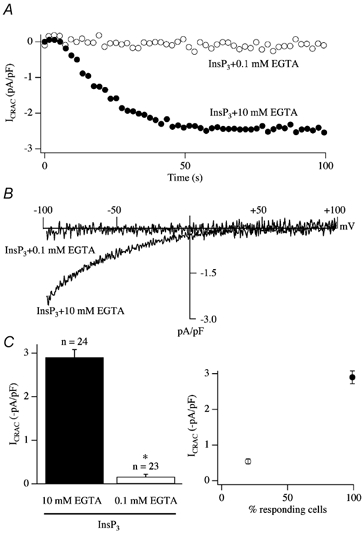
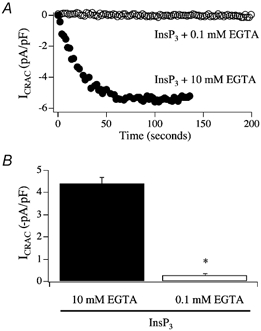
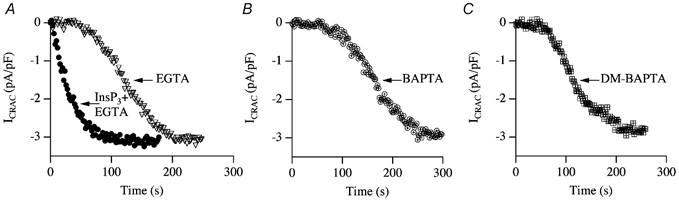
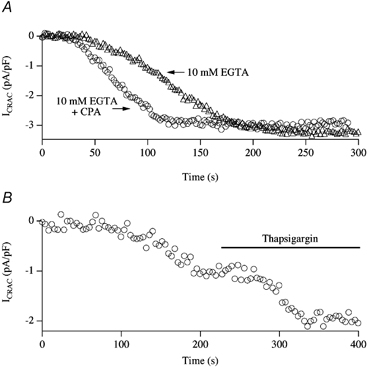
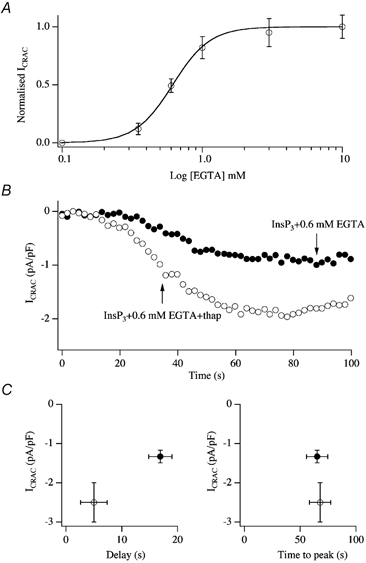
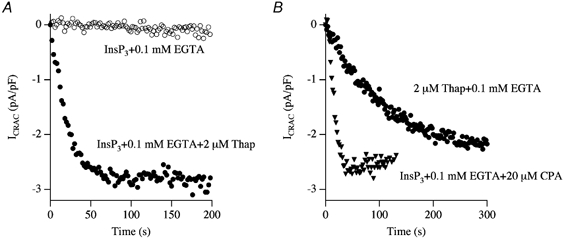
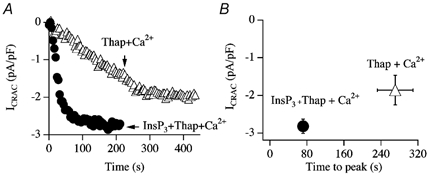
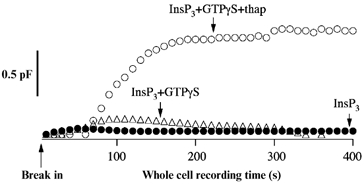
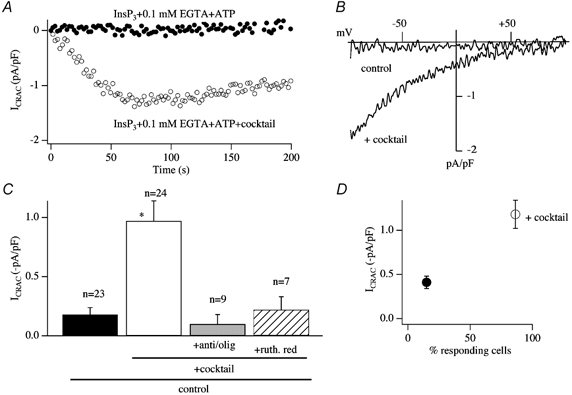
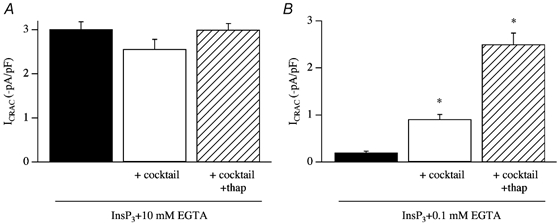
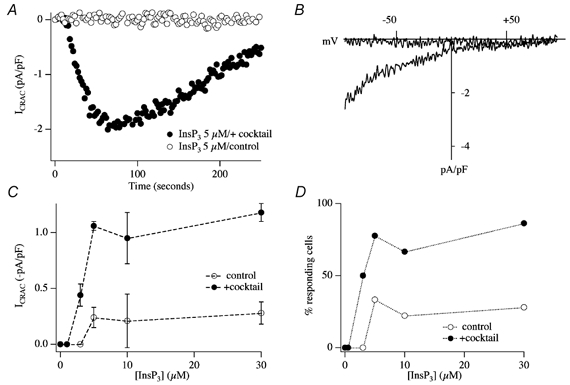
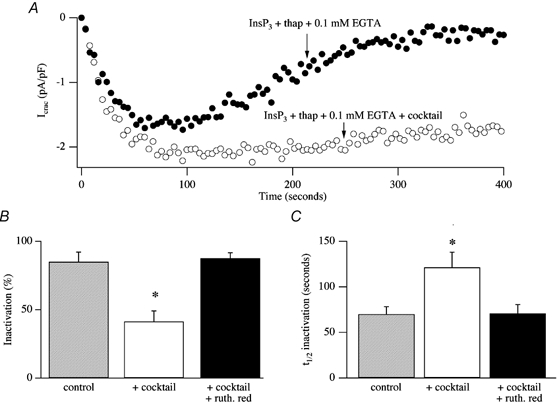
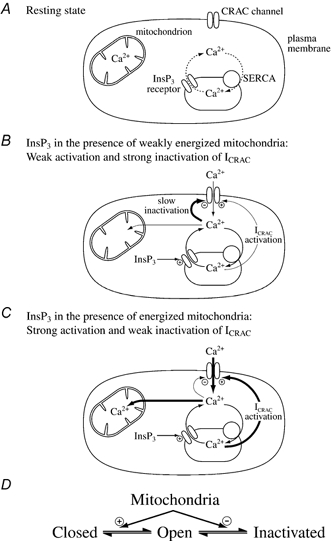
In eukaryotic cells, hormones and neurotransmitters that engage the phosphoinositide pathway evoke a biphasic increase in intracellular free Ca2+ concentration: an initial transient release of Ca2+ from intracellular stores is followed by a sustained phase of Ca2+ influx. This influx is generally store-dependent and is required for controlling a host of Ca2+-dependent processes ranging from exocytosis to cell growth and proliferation. In many cell types, store-operated Ca2+ entry is manifest as a non-voltage-gated Ca2+ current called ICRAC (Ca2+ release-activated Ca2+ current). Just how store emptying activates CRAC channels remains unclear, and some of our recent experiments that address this issue will be described. No less important from a physiological perspective is the weak Ca2+ buffer paradox: whereas macroscopic (whole cell) ICRAC can be measured routinely in the presence of strong intracellular Ca2+ buffer, the current is generally not detectable under physiological conditions of weak buffering following store emptying with the second messenger InsP3. In this review, I describe some of our experiments aimed at understanding just why InsP3 is ineffective under these conditions and which lead us to conclude that respiring mitochondria are essential for the activation of ICRAC in weak intracellular Ca2+ buffer. Mitochondrial Ca2+ uptake also increases the dynamic range over which InsP3 functions as the second messenger that controls Ca2+ influx. Finally, we find that Ca2+-dependent slow inactivation of Ca2+ influx, a widespread but poorly understood phenomenon that helps shape the profile of an intracellular Ca2+ signal, is regulated by mitochondrial Ca2+ buffering. Thus, by enabling macroscopic store-operated Ca2+ current to activate and then by controlling its extent and duration, mitochondria play a crucial role in all stages of store-operated Ca2+ influx. Store-operated Ca2+ entry reflects therefore a dynamic interplay between endoplasmic reticulum, mitochondria and plasma membrane.
Figures
References
-
- Alderton JM, Ahmed SA, Smith LA, Steinhardt RA. Evidence for a vesicle-mediated maintenance of store-operated calcium channels in a human embryonic kidney cell line. Cell Calcium. 2000;28:161–169. - PubMed
-
- Arnaudeau S, Kelley WL, Walsh JV, Demaurex N. Mitochondria recycle Ca2+ to the endoplasmic reticulum and prevent the depletion of neighbouring endoplasmic reticulum regions. J Biol Chem. 2001;276:29430–29439. - PubMed
-
- Artalejo AR, Ellory JC, Parekh AB. Ca2+ -dependent capacitance increases in rat basophilic leukemia cells following activation of store-operated Ca2+ entry and dialysis with high-Ca2+-containing intracellular solution. Pflugers Arch. 1998;436:934–939. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous