

A mechanism converting psychosocial stress into mononuclear cell activation
- PMID: 12578963
- PMCID: PMC149934
- DOI: 10.1073/pnas.0438019100
A mechanism converting psychosocial stress into mononuclear cell activation
Abstract
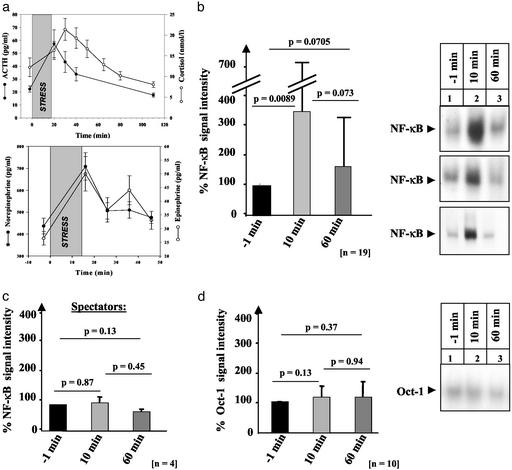
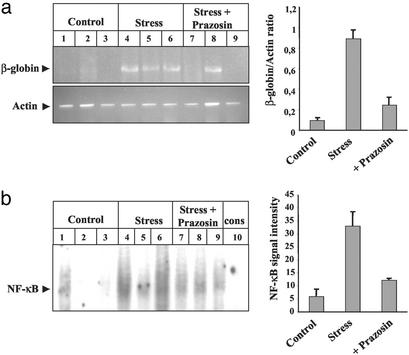
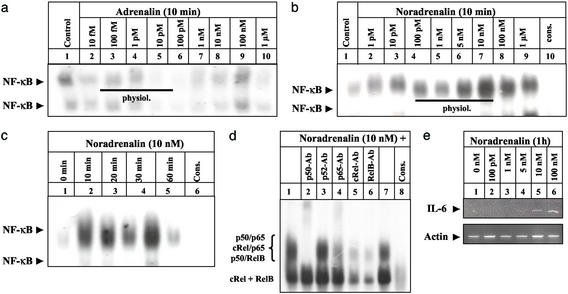
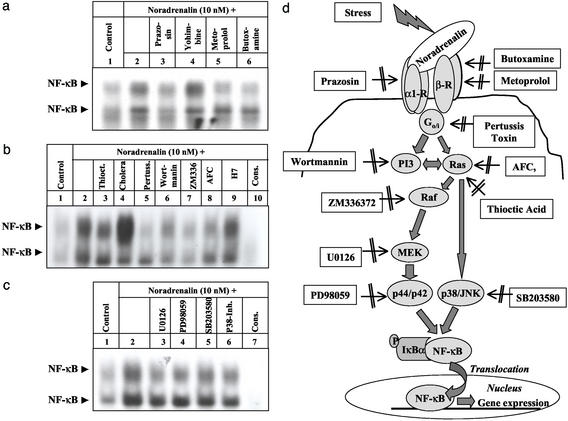
Little is known about the mechanisms converting psychosocial stress into cellular dysfunction. Various genes, up-regulated in atherosclerosis but also by psychosocial stress, are controlled by the transcription factor nuclear factor kappaB (NF-kappaB). Therefore, NF-kappaB is a good candidate to convert psychosocial stress into cellular activation. Volunteers were subjected to a brief laboratory stress test and NF-kappaB activity was determined in peripheral blood mononuclear cells (PBMC), as a window into the body and because PBMC play a role in diseases such as atherosclerosis. In 17 of 19 volunteers, NF-kappaB was rapidly induced during stress exposure, in parallel with elevated levels of catecholamines and cortisol, and returned to basal levels within 60 min. To model this response, mice transgenic for a strictly NF-kappaB-controlled beta-globin transgene were stressed by immobilization. Immobilization resulted in increased beta-globin expression, which could be reduced in the presence of the alpha1-adrenergic inhibitor prazosin. To define the role of adrenergic stimulation in the up-regulation of NF-kappaB, THP-1 cells were induced with physiological amounts of catecholamines for 10 min. Only noradrenaline resulted in a dose- and time-dependent induction of NF-kappaB and NF-kappaB-dependent gene expression, which depended on pertussis-toxin-sensitive G protein-mediated phosphophatidylinositol 3-kinase, Ras/Raf, and mitogen-activated protein kinase activation. Induction was reduced by alpha(1)- and beta-adrenergic inhibitors. Thus, noradrenaline-dependent adrenergic stimulation results in activation of NF-kappaB in vitro and in vivo. Activation of NF-kappaB represents a downstream effector for the neuroendocrine response to stressful psychosocial events and links changes in the activity of the neuroendocrine axis to the cellular response.
Figures
References
-
- Rozanski A, Blumenthal J A, Kaplan J. Circulation. 2000;99:2192–2217. - PubMed
-
- Leor J, Poole W K, Kloner R A. N Engl J Med. 1996;334:413–419. - PubMed
-
- Meisel S R, Kutz I, Dayan K I, Pauzner H, Chetboun I, Arbel Y, David D. Lancet. 1991;338:660–661. - PubMed
-
- Stansfeld S A, Fuhrer R, Shipley M J, Marmot M G. Int J Epidemiol. 2002;31:248–255. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
