

Why study human limb malformations?
- PMID: 12587917
- PMCID: PMC1571054
- DOI: 10.1046/j.1469-7580.2003.00130.x
Why study human limb malformations?
Abstract
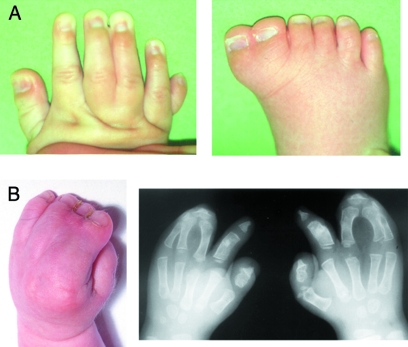
Congenital limb malformations occur in 1 in 500 to 1 in 1000 human live births and include both gross reduction defects and more subtle alterations in the number, length and anatomy of the digits. The major causes of limb malformations are abnormal genetic programming and intra-uterine disruption to development. The identification of causative gene mutations is important for genetic counselling and also provides insights into the mechanisms controlling limb development. This article illustrates some of the lessons learnt from the study of human limb malformation, organized into seven categories. These are: (1) identification of novel genes, (2) allelic mutation series, (3) pleiotropy, (4) qualitative or (5) quantitative differences between mouse and human development, (6) physical and teratogenic disruption, and (7) unusual biological phenomena.
Figures



References
-
- Afzal AR, Rajab A, Fenske CD, Oldridge M, Elanko N, Ternes-Pereira E, et al. Recessive Robinow syndrome, allelic to dominant brachydactyly type B, is caused by mutation of ROR2. Nature Genet. 2000;25:419–422. - PubMed
-
- Anderson J, Burns HD, Enriquez-Harris P, Wilkie AOM, Heath JK. Apert syndrome mutations in fibroblast growth factor receptor 2 exhibit increased affinity for FGF ligand. Hum. Mol. Genet. 1998;7:1475–1483. - PubMed
-
- Ballabio A. The rise and fall of positional cloning? Nature Genet. 1993;3:277–279. - PubMed
-
- Biesecker LG. Genotype-phenotype correlation in human GLI3 disorders. Eur. J. Hum. Genet. 2001;9(Suppl. 1):76.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
