

Mutations in genes encoding fast-twitch contractile proteins cause distal arthrogryposis syndromes
- PMID: 12592607
- PMCID: PMC1180243
- DOI: 10.1086/368294
Mutations in genes encoding fast-twitch contractile proteins cause distal arthrogryposis syndromes
Abstract
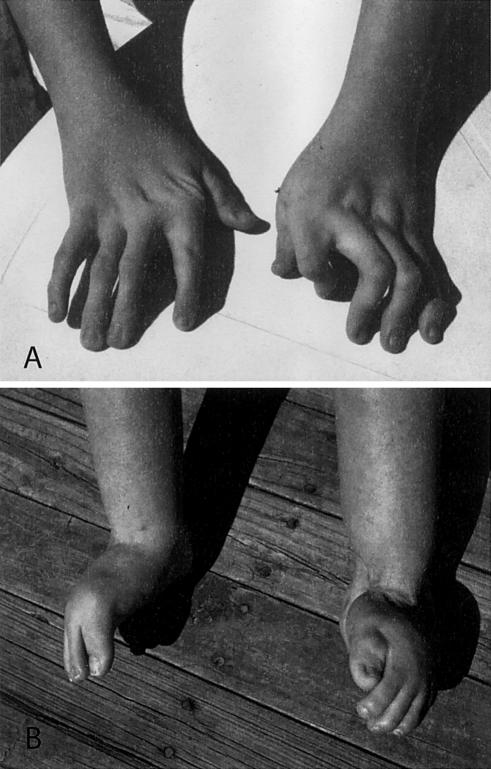
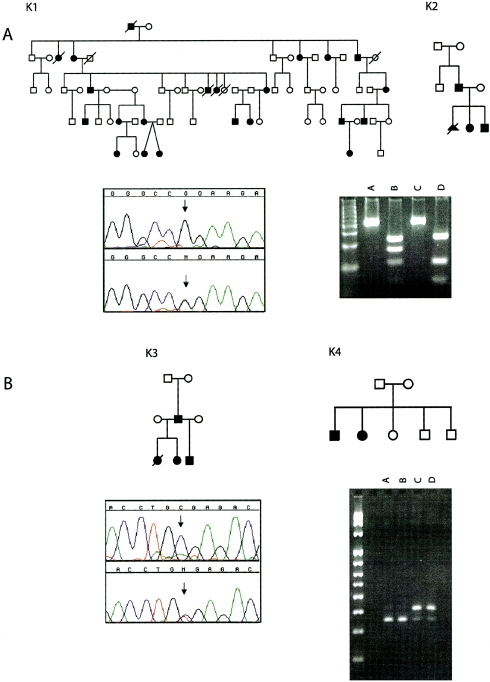
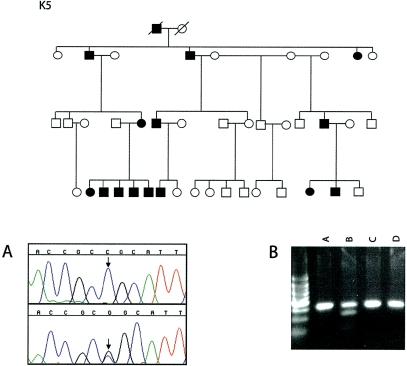
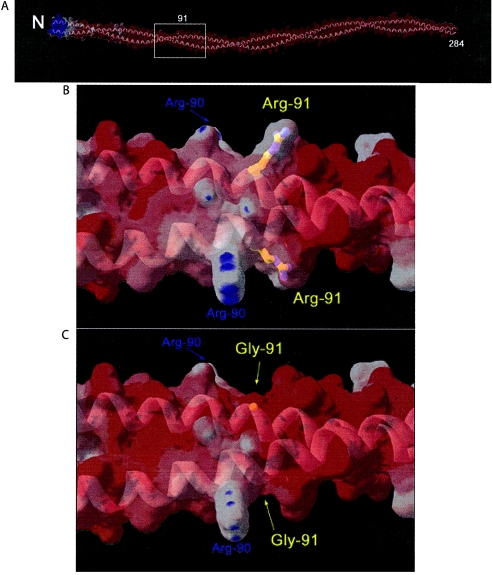
The distal arthrogryposes (DAs) are a group of disorders characterized by multiple congenital contractures of the limbs. We previously mapped a locus for DA type 2B (DA2B), the most common of the DAs, to chromosome 11. We now report that DA2B is caused by mutations in TNNI2 that are predicted to disrupt the carboxy-terminal domain of an isoform of troponin I (TnI) specific to the troponin-tropomyosin (Tc-Tm) complex of fast-twitch myofibers. Because the DAs are genetically heterogeneous, we sought additional candidate genes by examining modifiers of mutant Drosophila isoforms of TnI. One of these modifiers, Tm2, encodes tropomyosin, another component of the Tc-Tm complex. A human homologue of Tm2, TPM2, encodes beta-tropomyosin and maps to the critical interval of DA type 1 (DA1). We discovered that DA1 is caused by substitution of a highly conserved amino acid residue in beta-tropomyosin. These findings suggest that DAs, in general, may be caused by mutations in genes encoding proteins of the contractile apparatus specific to fast-twitch myofibers. This provides a new opportunity to directly study the etiology and pathogenesis of multiple-congenital-contracture syndromes.
Figures


References
Electronic-Database Information
-
- Online Mendelian Inheritance in Man (OMIM) http://www.ncbi.nlm.nih.gov/Omim/ (for DA1 [MIM 108120], DA2B [MIM 601680], and FSS [MIM 193700])
References
-
- Andrade MD, Barnholtz JS, Amos CI, Lockmiller C, Scott A, Risman M, Hecht J (1998) Segregation analysis of idiopathic talipes equinovarus in a Texan population. Am J Med Genet 79:97–102 - PubMed
-
- Bamshad M, Bohnsack JF, Jorde LB, Carey JC (1996a) Distal arthrogryposis type 1: clinical analysis of a large kindred. Am J Med Genet 65:282–285 - PubMed
-
- Bamshad M, Jorde LB, Carey JC (1996b) A revised and extended classification of the distal arthrogryposes. Am J Med Genet 65:277–281 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
