

The structure of full-length LysR-type transcriptional regulators. Modeling of the full-length OxyR transcription factor dimer
- PMID: 12595552
- PMCID: PMC149827
- DOI: 10.1093/nar/gkg234
The structure of full-length LysR-type transcriptional regulators. Modeling of the full-length OxyR transcription factor dimer
Abstract
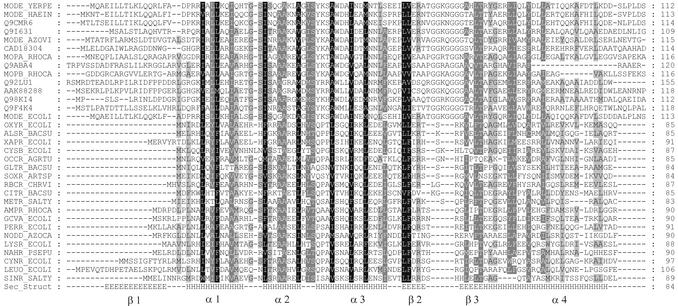
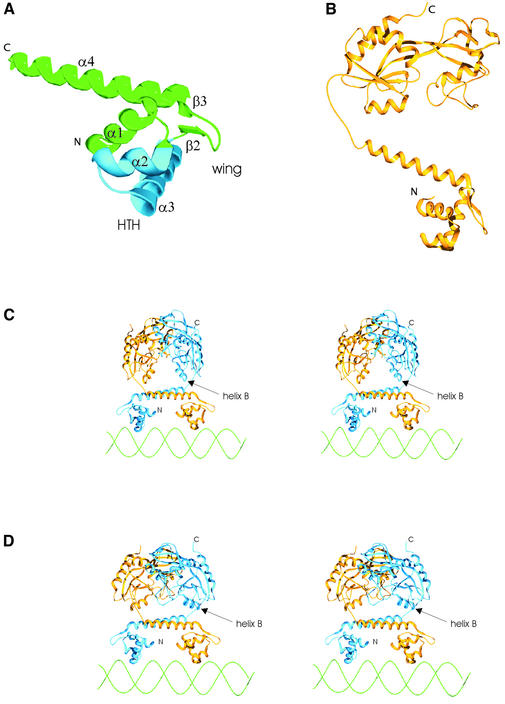
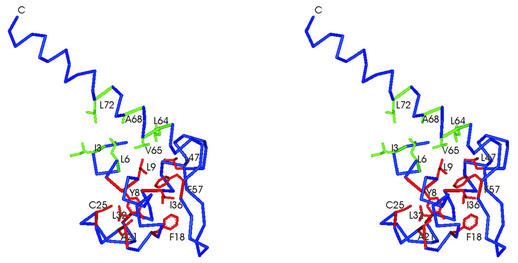
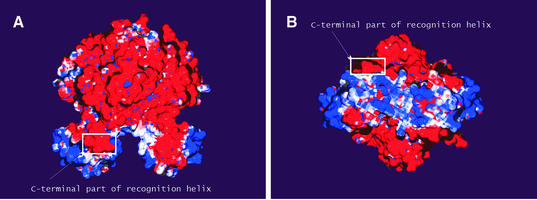
The LysR-type transcriptional regulators (LTTRs) comprise the largest family of prokaryotic transcription factors. These proteins are composed of an N-terminal DNA binding domain (DBD) and a C-terminal cofactor binding domain. To date, no structure of the DBD has been solved. According to the SUPERFAMILY and MODBASE databases, a reliable homology model of LTTR DBDs may be built using the structure of the Escherichia coli ModE transcription factor, containing a winged helix- turn-helix (HTH) motif, as a template. The remote, but statistically significant, sequence similarity between ModE and LTTR DBDs and an alignment generated using SUPERFAMILY and MODBASE methods was independently confirmed by alignment of sequence profiles representing ModE and LTTR family DBDs. Using the crystal structure of the E.coli OxyR C-terminal domain and the DBD alignments we constructed a structural model of the full-length dimer of this LTTR family member and used it to investigate the mode of protein-DNA interaction. We also applied the model to interpret, in a structural context, the results of numerous biochemical studies of mutated LTTRs. A comparison of the LTTR DBD model with the structures of other HTH proteins also provides insights into the interaction of LTTRs with the C-terminal domain of the RNA polymerase alpha subunit.
Figures
Similar articles
-
Structural and functional insights into transcription activation of the essential LysR-type transcriptional regulators.Protein Sci. 2024 Jun;33(6):e5012. doi: 10.1002/pro.5012. Protein Sci. 2024. PMID: 38723180 Free PMC article.
-
Crystal structure of ArgP from Mycobacterium tuberculosis confirms two distinct conformations of full-length LysR transcriptional regulators and reveals its function in DNA binding and transcriptional regulation.J Mol Biol. 2010 Mar 5;396(4):1012-24. doi: 10.1016/j.jmb.2009.12.033. Epub 2009 Dec 28. J Mol Biol. 2010. PMID: 20036253
-
Crystal structure of a full-length LysR-type transcriptional regulator, CbnR: unusual combination of two subunit forms and molecular bases for causing and changing DNA bend.J Mol Biol. 2003 May 2;328(3):555-66. doi: 10.1016/s0022-2836(03)00312-7. J Mol Biol. 2003. PMID: 12706716
-
Structure and function of the LysR-type transcriptional regulator (LTTR) family proteins.Microbiology (Reading). 2008 Dec;154(Pt 12):3609-3623. doi: 10.1099/mic.0.2008/022772-0. Microbiology (Reading). 2008. PMID: 19047729 Review.
-
Molecular biology of the LysR family of transcriptional regulators.Annu Rev Microbiol. 1993;47:597-626. doi: 10.1146/annurev.mi.47.100193.003121. Annu Rev Microbiol. 1993. PMID: 8257110 Review.
Cited by
-
Whole-genome sequencing and phenotypic analysis of Bacillus subtilis mutants following evolution under conditions of relaxed selection for sporulation.Appl Environ Microbiol. 2011 Oct;77(19):6867-77. doi: 10.1128/AEM.05272-11. Epub 2011 Aug 5. Appl Environ Microbiol. 2011. PMID: 21821766 Free PMC article.
-
Structural and functional insights into transcription activation of the essential LysR-type transcriptional regulators.Protein Sci. 2024 Jun;33(6):e5012. doi: 10.1002/pro.5012. Protein Sci. 2024. PMID: 38723180 Free PMC article.
-
A LysR Transcriptional Regulator Manipulates Macrophage Autophagy Flux During Brucella Infection.Front Cell Infect Microbiol. 2022 Mar 22;12:858173. doi: 10.3389/fcimb.2022.858173. eCollection 2022. Front Cell Infect Microbiol. 2022. PMID: 35392609 Free PMC article.
-
SstF, a novel sulforaphane-sensing transcription factor of Xanthomonas campestris, is required for sulforaphane tolerance and virulence.Mol Plant Pathol. 2023 May;24(5):452-465. doi: 10.1111/mpp.13314. Epub 2023 Feb 24. Mol Plant Pathol. 2023. PMID: 36829260 Free PMC article.
-
How Bacterial Redox Sensors Transmit Redox Signals via Structural Changes.Antioxidants (Basel). 2021 Mar 24;10(4):502. doi: 10.3390/antiox10040502. Antioxidants (Basel). 2021. PMID: 33804871 Free PMC article. Review.
References
-
- Schell M.A. (1993) Molecular biology of the LysR family of transcriptional regulators. Annu. Rev. Microbiol., 47, 597–626. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
