

Common molecular determinants of flecainide and lidocaine block of heart Na+ channels: evidence from experiments with neutral and quaternary flecainide analogues
- PMID: 12601084
- PMCID: PMC2217334
- DOI: 10.1085/jgp.20028723
Common molecular determinants of flecainide and lidocaine block of heart Na+ channels: evidence from experiments with neutral and quaternary flecainide analogues
Abstract
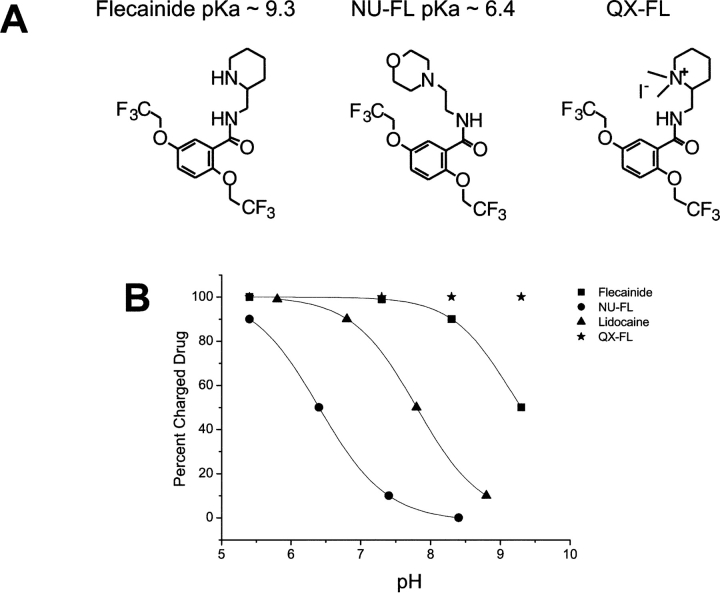
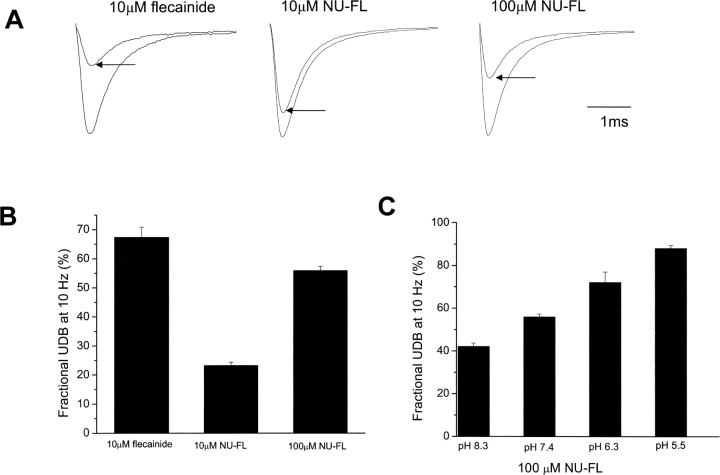
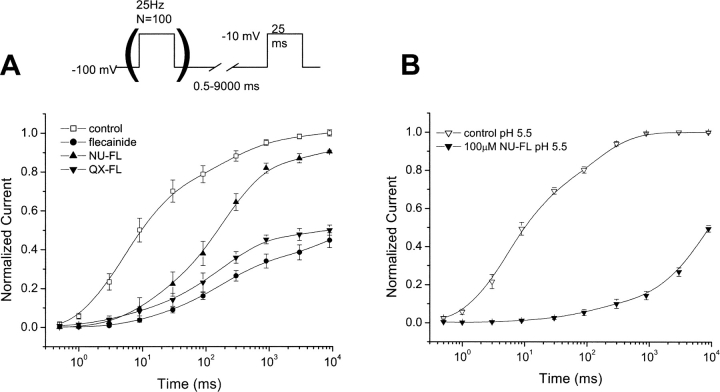
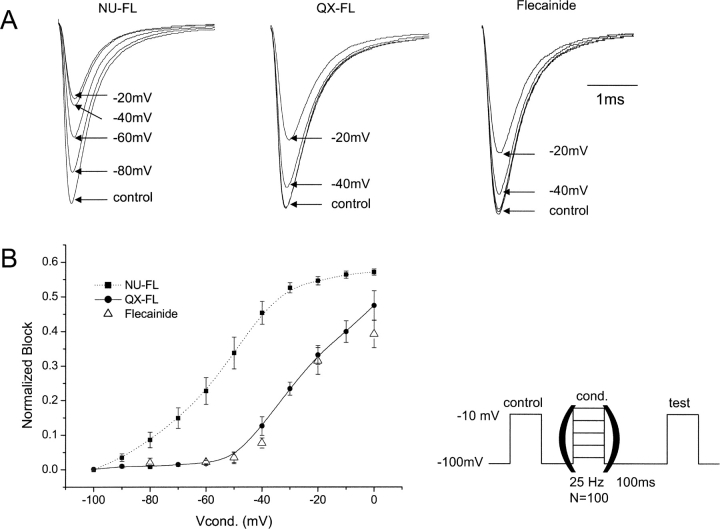
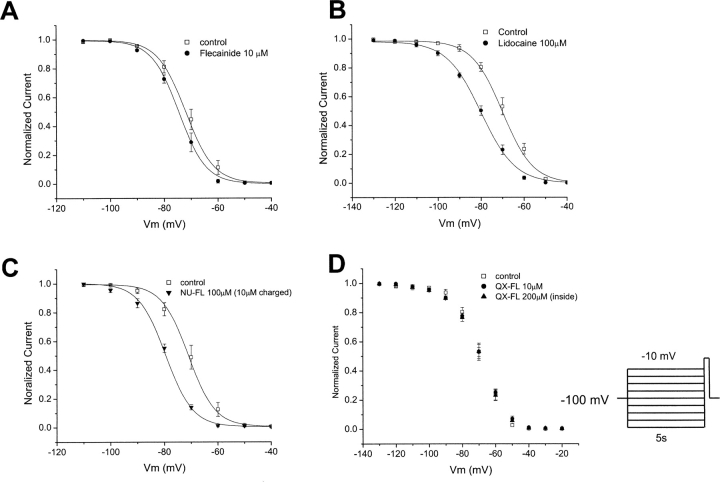
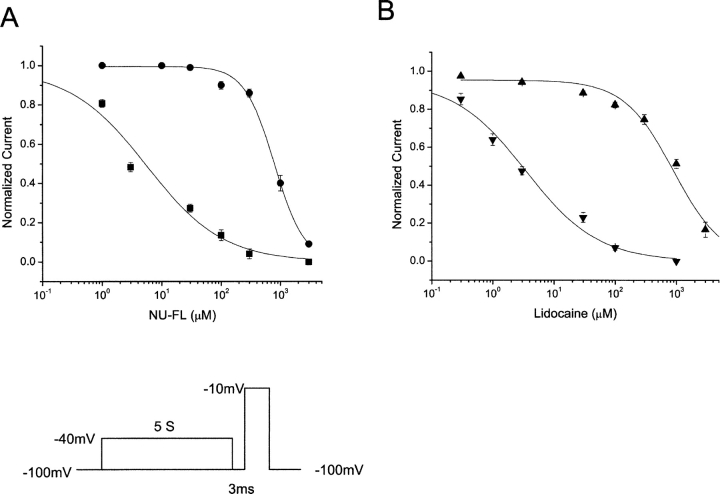
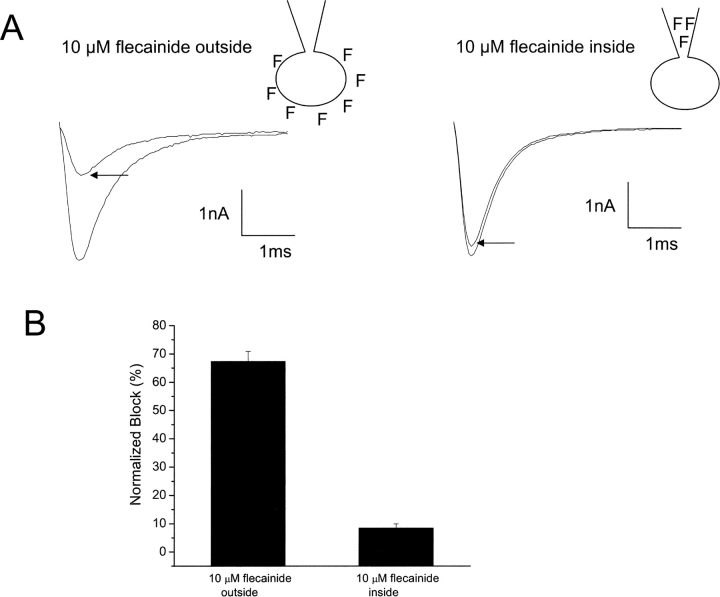
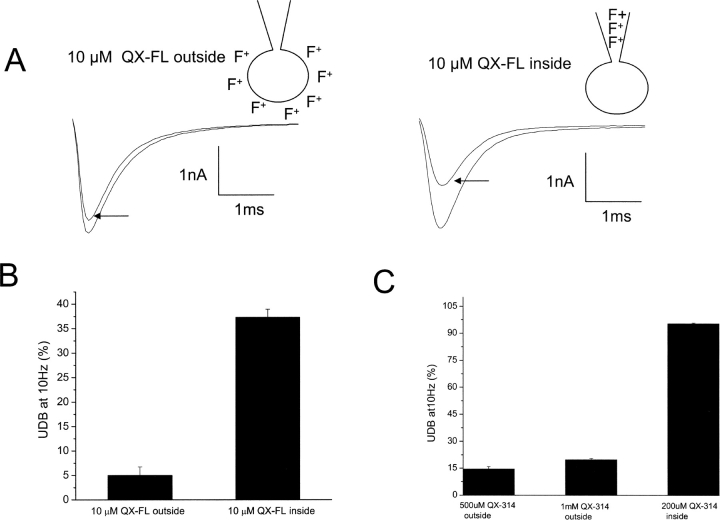
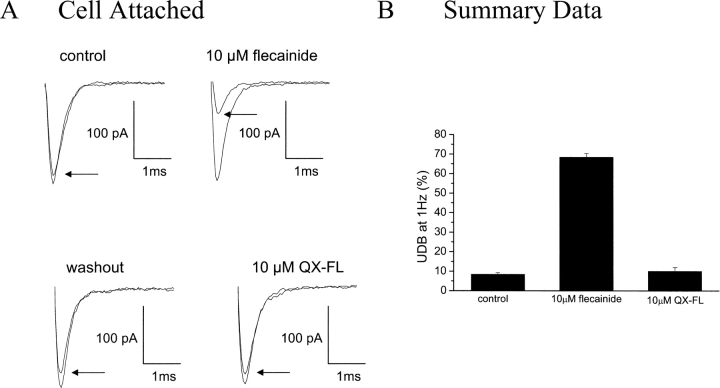
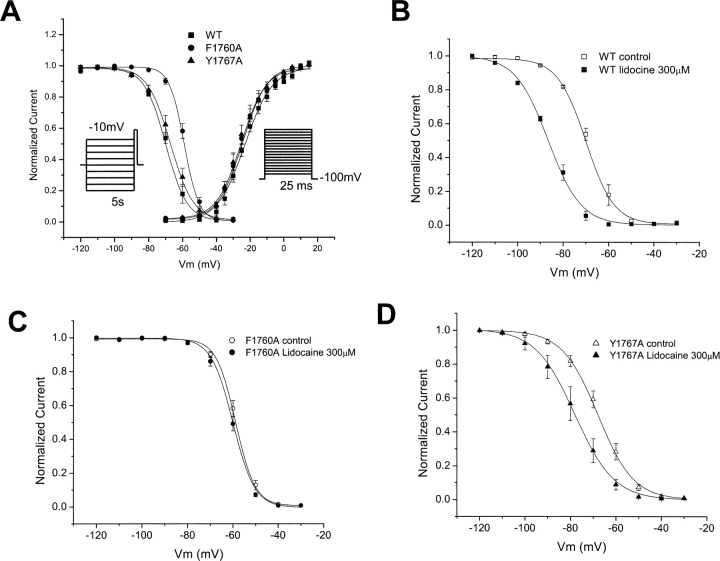
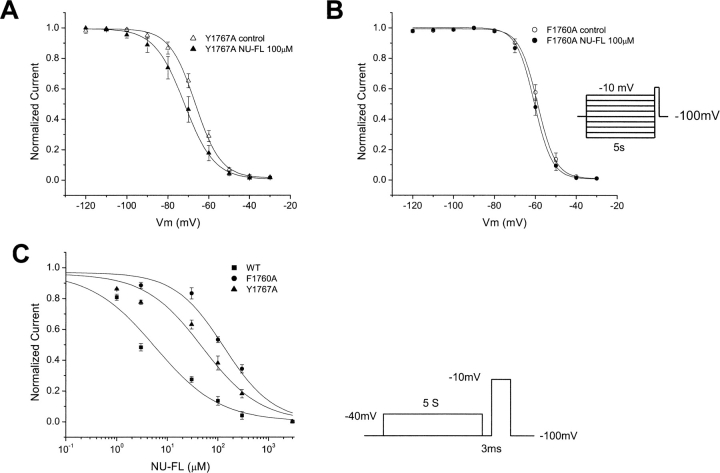
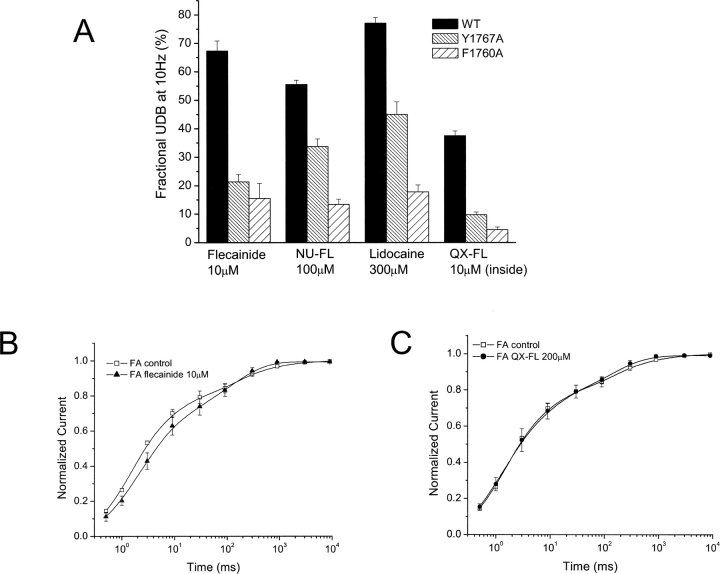
Flecainide (pKa 9.3, 99% charged at pH 7.4) and lidocaine (pKa 7.6-8.0, approximately 50% neutral at pH 7.4) have similar structures but markedly different effects on Na(+) channel activity. Both drugs cause well-characterized use-dependent block (UDB) of Na(+) channels due to stabilization of the inactivated state, but flecainide requires that channels first open before block develops, whereas lidocaine is believed to bind directly to the inactivated state. To test whether the charge on flecainide might determine its state specificity of Na(+) channel blockade, we developed two flecainide analogues, NU-FL (pKa 6.4), that is 90% neutral at pH 7.4, and a quaternary flecainide analogue, QX-FL, that is fully charged at physiological pH. We examined the effects of flecainide, NU-FL, QX-FL, and lidocaine on human cardiac Na(+) channels expressed in human embryonic kidney (HEK) 293 cells. At physiological pH, NU-FL, like lidocaine but not flecainide, interacts preferentially with inactivated channels without prerequisite channel opening, and causes minimal UDB. We find that UDB develops predominantly by the charged form of flecainide as evidenced by investigation of QX-FL at physiological pH and NU-FL investigated over a more acidic pH range where its charged fraction is increased. QX-FL is a potent blocker of channels when applied from inside the cell, but acts very weakly with external application. UDB by QX-FL, like flecainide, develops only after channels open. Once blocked, channels recover very slowly from QX-FL block, apparently without requisite channel opening. Our data strongly suggest that it is the difference in degree of ionization (pKa) between lidocaine and flecainide, rather than gross structural features, that determines distinction in block of cardiac Na(+) channels. The data also suggest that the two drugs share a common receptor but, consistent with the modulated receptor hypothesis, reach this receptor by distinct routes dictated by the degree of ionization of the drug molecules.
Figures
References
-
- Abriel, H., C. Cabo, X.H. Wehrens, I. Rivolta, H.K. Motoike, M. Memmi, C. Napolitano, S.G. Priori, and R.S. Kass. 2001. Novel arrhythmogenic mechanism revealed by a long-qt syndrome mutation in the cardiac na(+) channel. Circ. Res. 88:740–745. - PubMed
-
- An, R.H., R. Bangalore, S.Z. Rosero, and R.S. Kass. 1996. Lidocaine block of LQT-3 mutant human Na+ channels. Circ. Res. 79:103–108. - PubMed
-
- Anno, T., and L.M. Hondeghem. 1990. Interactions of flecainide with guinea pig cardiac sodium channels. Importance of activation unblocking to the voltage dependence of recovery. Circ. Res. 66:789–803. - PubMed
-
- Banitt, E.H., W.E. Coyne, J.R. Schmid, and A. Mendel. 1975. Antiarrhythmics. N-(aminoalkylene)trifluoroethoxybenzamides and N-(aminoalkylene)trifluoroethoxynaphthamides. J. Med. Chem. 18:1130–1134. - PubMed
-
- Benhorin, J., R. Taub, M. Goldmit, B. Kerem, R.S. Kass, I. Windman, and A. Medina. 2000. Effects of flecainide in patients with new SCN5A mutation: mutation- specific therapy for long-QT syndrome? Circulation. 101:1698–1706. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
