

Molecular dynamics studies of the wild-type and double mutant HIV-1 integrase complexed with the 5CITEP inhibitor: mechanism for inhibition and drug resistance
- PMID: 12609852
- PMCID: PMC1302719
- DOI: 10.1016/S0006-3495(03)74958-3
Molecular dynamics studies of the wild-type and double mutant HIV-1 integrase complexed with the 5CITEP inhibitor: mechanism for inhibition and drug resistance
Abstract
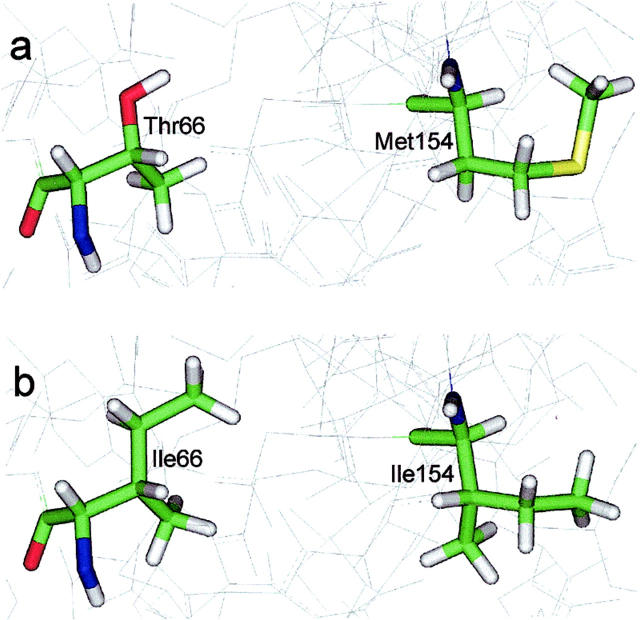
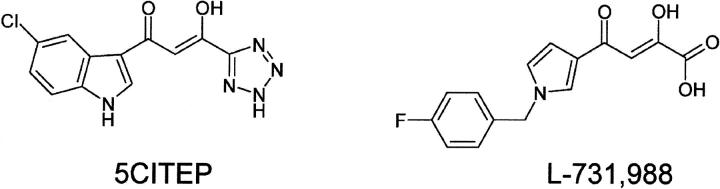
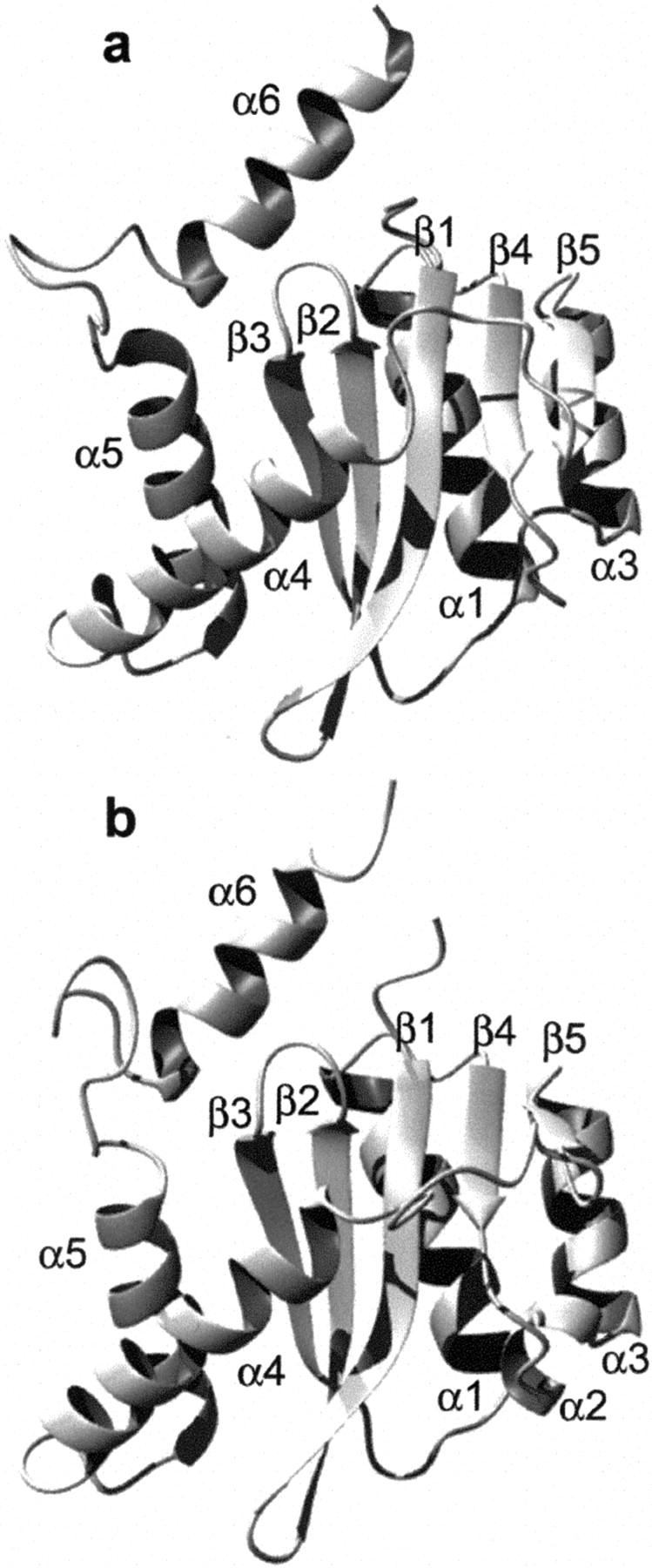
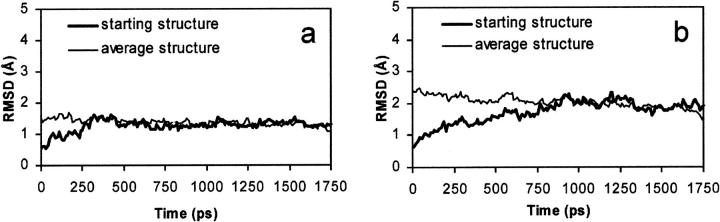
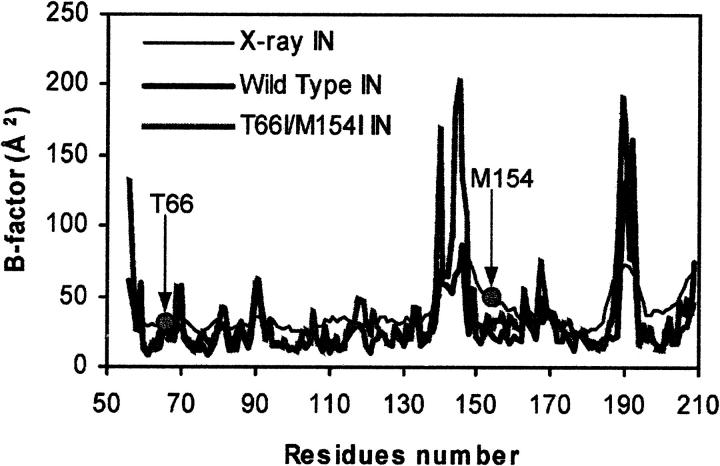
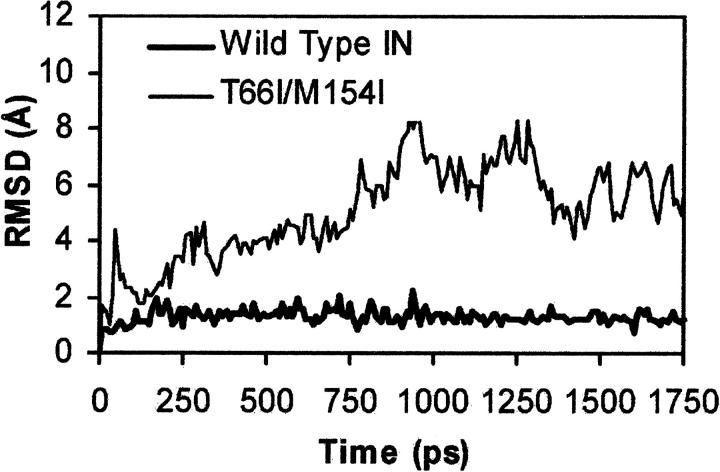
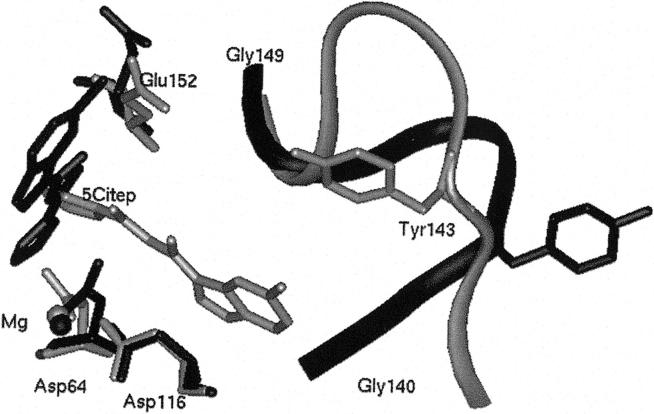
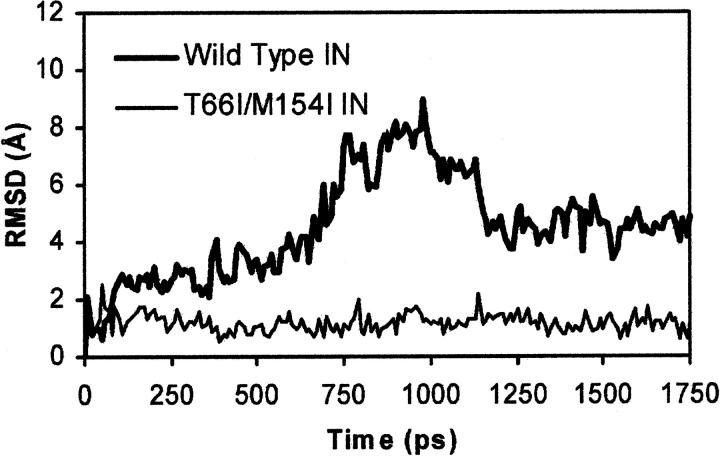
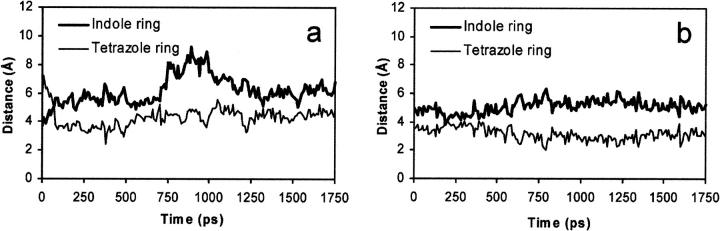
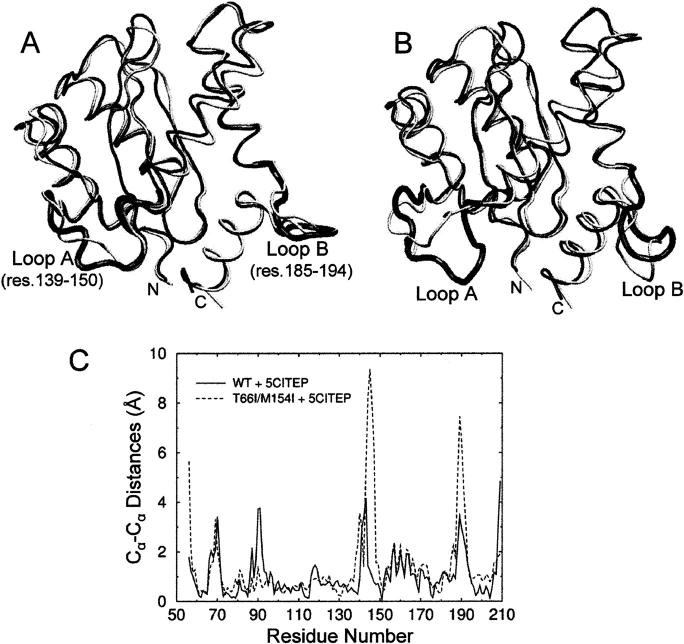
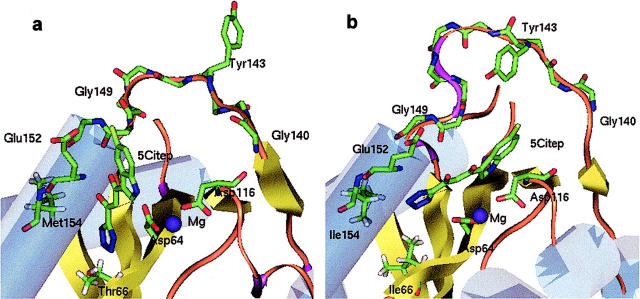
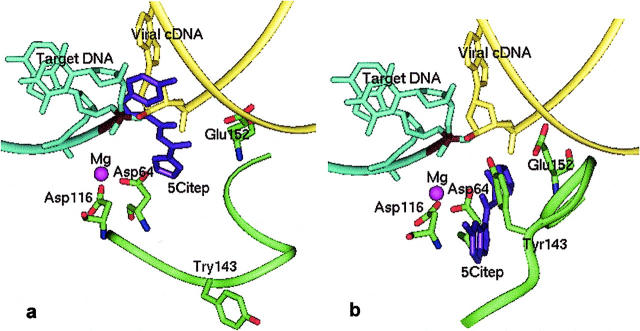
The human immunodeficiency virus type 1 (HIV-1) integrase (IN) is an essential enzyme in the life cycle of the virus and is an attractive target for the development of new drugs useful in acquired immunodeficiency syndrome multidrug therapy. Starting from the crystal structure of the 5CITEP inhibitor bound to the active site in the catalytic domain of the HIV-1 IN, two different molecular dynamics simulations in water have been carried out. In the first simulation the wild-type IN was used, whereas in the second one the double mutation T66I/M154I, described to lead to drug resistance, was introduced in the protein. Compelling differences have been observed in these two structures during analyses of the molecular dynamics trajectories, particularly in the inhibitor binding modes and in the conformational flexibility of the loop (residues 138-149) located near the three catalytic residues in the active site (Asp(64), Asp(116), Glu(152)). Because the conformational flexibility of this region is important for efficient biological activity and its behavior is quite different in the two models, we suggest a hypothetical mechanism for the inhibition and drug resistance of HIV-1 IN. These results can be useful for the rational design of more potent and selective integrase inhibitors and may allow for the design of inhibitors that will be more robust against known resistance mutations.
Figures
Similar articles
-
Comparative molecular dynamics simulations of HIV-1 integrase and the T66I/M154I mutant: binding modes and drug resistance to a diketo acid inhibitor.Proteins. 2005 Jun 1;59(4):723-41. doi: 10.1002/prot.20447. Proteins. 2005. PMID: 15815973
-
Comparison of multiple molecular dynamics trajectories calculated for the drug-resistant HIV-1 integrase T66I/M154I catalytic domain.Biophys J. 2005 May;88(5):3072-82. doi: 10.1529/biophysj.104.050286. Epub 2005 Mar 11. Biophys J. 2005. PMID: 15764656 Free PMC article.
-
Ordered water and ligand mobility in the HIV-1 integrase-5CITEP complex: a molecular dynamics study.J Med Chem. 2001 Sep 13;44(19):3043-7. doi: 10.1021/jm010205y. J Med Chem. 2001. PMID: 11543671
-
Characterization and structural analysis of HIV-1 integrase conservation.AIDS Rev. 2009 Jan-Mar;11(1):17-29. AIDS Rev. 2009. PMID: 19290031 Review.
-
HIV-1 integrase: the next target for AIDS therapy?Pathol Biol (Paris). 2001 Apr;49(3):237-46. doi: 10.1016/s0369-8114(01)00135-3. Pathol Biol (Paris). 2001. PMID: 11367559 Review.
Cited by
-
Evolutionary basis for the coupled-domain motions in Thermus thermophilus leucyl-tRNA synthetase.J Biol Chem. 2009 Apr 10;284(15):10088-99. doi: 10.1074/jbc.M807361200. Epub 2009 Feb 2. J Biol Chem. 2009. PMID: 19188368 Free PMC article.
-
Exploring the binding of HIV-1 integrase inhibitors by comparative residue interaction analysis (CoRIA).J Mol Model. 2009 Mar;15(3):233-45. doi: 10.1007/s00894-008-0399-4. Epub 2008 Dec 2. J Mol Model. 2009. PMID: 19048312
-
Molecular dynamics: survey of methods for simulating the activity of proteins.Chem Rev. 2006 May;106(5):1589-615. doi: 10.1021/cr040426m. Chem Rev. 2006. PMID: 16683746 Free PMC article. Review. No abstract available.
-
Design and synthesis of bis-amide and hydrazide-containing derivatives of malonic acid as potential HIV-1 integrase inhibitors.Molecules. 2008 Oct 1;13(10):2442-61. doi: 10.3390/molecules13102442. Molecules. 2008. PMID: 18830166 Free PMC article.
-
Human immunodeficiency virus type 1 (HIV-1) integrase: resistance to diketo acid integrase inhibitors impairs HIV-1 replication and integration and confers cross-resistance to L-chicoric acid.J Virol. 2004 Jun;78(11):5835-47. doi: 10.1128/JVI.78.11.5835-5847.2004. J Virol. 2004. PMID: 15140981 Free PMC article.
References
-
- Abagyan, R. A., M. M. Totrov, and D. N. Kuznetsov. 1994. ICM – a new method for protein modeling and design. Applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 15:488–506.
-
- Amadei, A., A. B. Linssen, and H. J. Berendsen. 1993. Essential dynamics of proteins. Proteins. 17:412–425. - PubMed
-
- Anchell, J., E. Apra, D. Bernholdt, P. Borowski, E. Bylaska, T. Clark, D. Clerc, H. Dachsel, W. de Jong, M. Deegan, M. Dupuis, K. Dyall, D. Elwood, G. Fann, H. Fruchtl, E. Glendenning, M. Gutowski, R. Harrison, A. Hess, J. Jaffe, B. Johnson, J. Ju, R. Kendall, R. Kobayashi, R. Kutteh, Z. Lin, R. Littlefield, X. Long, B. Meng, J. Nichols, J. Nieplocha, A. Rendall, M. Rosing, G. Sandrone, M. Stave, T. P. Straatsma, H. Taylor, G. Thomas, J. van Lenthe, T. Windus, K. Wolinski, A. Wong, and Z. Zhang. 1999. NWChem, A Computational Chemistry Package for Parallel Computers, Version 3.3.1. High Performance Computational Chemistry Group, Pacific Northwestern National Laboratory, Richland, WA.
-
- Asante-Appiah, E., and A. M. Skalka. 1999. HIV-1 integrase: structural organization, conformational changes, and catalysis. Adv. Virus Res. 52:351–369. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
