

Protein structure and dynamics in nonaqueous solvents: insights from molecular dynamics simulation studies
- PMID: 12609866
- PMCID: PMC1302733
- DOI: 10.1016/S0006-3495(03)74972-8
Protein structure and dynamics in nonaqueous solvents: insights from molecular dynamics simulation studies
Abstract
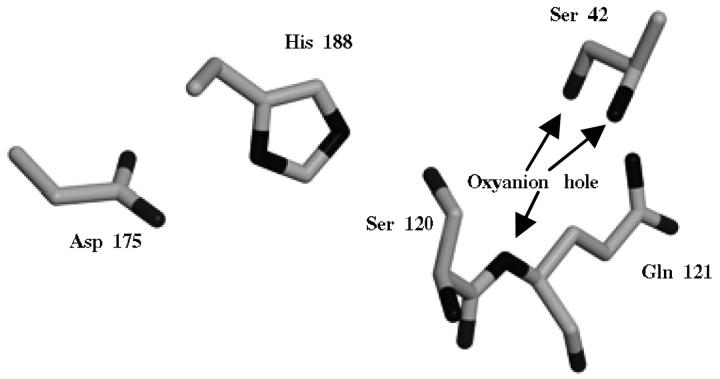
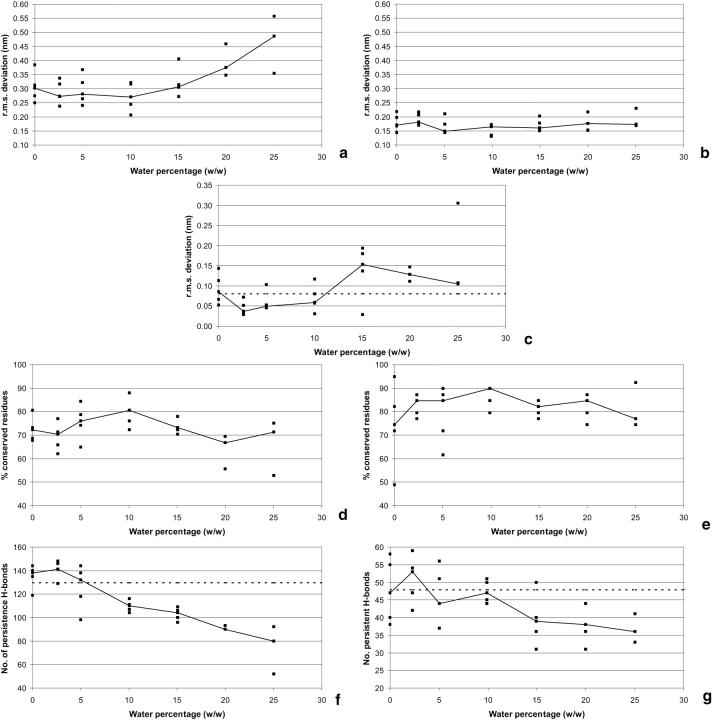
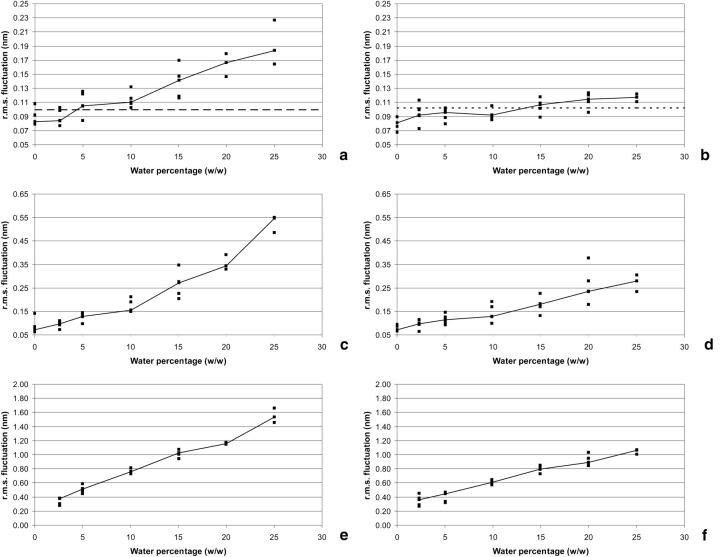
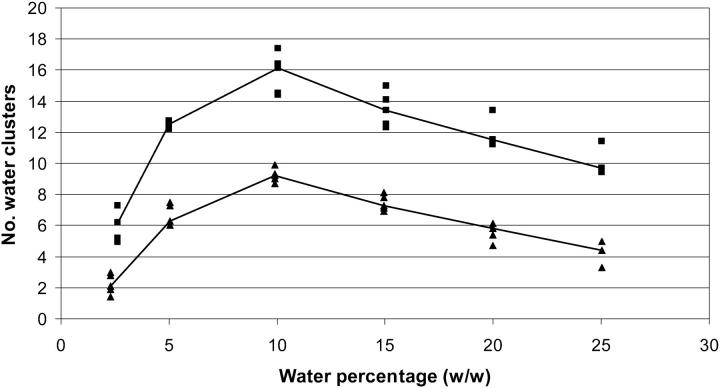
Protein structure and dynamics in nonaqueous solvents are here investigated using molecular dynamics simulation studies, by considering two model proteins (ubiquitin and cutinase) in hexane, under varying hydration conditions. Ionization of the protein groups is treated assuming "pH memory," i.e., using the ionization states characteristic of aqueous solution. Neutralization of charged groups by counterions is done by considering a counterion for each charged group that cannot be made neutral by establishing a salt bridge with another charged group; this treatment is more physically reasonable for the nonaqueous situation, contrasting with the usual procedures. Our studies show that hydration has a profound effect on protein stability and flexibility in nonaqueous solvents. The structure becomes more nativelike with increasing values of hydration, up to a certain point, when further increases render it unstable and unfolding starts to occur. There is an optimal amount of water, approximately 10% (w/w), where the protein structure and flexibility are closer to the ones found in aqueous solution. This behavior can explain the experimentally known bell-shaped dependence of enzyme catalysis on hydration, and the molecular reasons for it are examined here. Water and counterions play a fundamental and dynamic role on protein stabilization, but they also seem to be important for protein unfolding at high percentages of bound water.
Figures
Similar articles
-
Water dependent properties of cutinase in nonaqueous solvents: a computational study of enantioselectivity.Biophys J. 2005 Aug;89(2):999-1008. doi: 10.1529/biophysj.105.063297. Epub 2005 May 27. Biophys J. 2005. PMID: 15923226 Free PMC article.
-
Modeling hydration mechanisms of enzymes in nonpolar and polar organic solvents.FEBS J. 2007 May;274(9):2424-36. doi: 10.1111/j.1742-4658.2007.05781.x. FEBS J. 2007. PMID: 17419728
-
Hydration of enzyme in nonaqueous media is consistent with solvent dependence of its activity.Biophys J. 2004 Aug;87(2):812-21. doi: 10.1529/biophysj.104.041269. Biophys J. 2004. PMID: 15298890 Free PMC article.
-
The role of dynamics in enzyme activity.Annu Rev Biophys Biomol Struct. 2003;32:69-92. doi: 10.1146/annurev.biophys.32.110601.142445. Epub 2002 Dec 2. Annu Rev Biophys Biomol Struct. 2003. PMID: 12471064 Review.
-
Water mediation in protein folding and molecular recognition.Annu Rev Biophys Biomol Struct. 2006;35:389-415. doi: 10.1146/annurev.biophys.35.040405.102134. Annu Rev Biophys Biomol Struct. 2006. PMID: 16689642 Review.
Cited by
-
Long dynamics simulations of proteins using atomistic force fields and a continuum representation of solvent effects: calculation of structural and dynamic properties.Proteins. 2005 Aug 15;60(3):464-84. doi: 10.1002/prot.20470. Proteins. 2005. PMID: 15959866 Free PMC article.
-
Solvent-induced lid opening in lipases: a molecular dynamics study.Protein Sci. 2010 Nov;19(11):2122-30. doi: 10.1002/pro.493. Protein Sci. 2010. PMID: 20812327 Free PMC article.
-
Calcium-ion-induced stabilization of the protease from Bacillus cereus WQ9-2 in aqueous hydrophilic solvents: effect of calcium ion binding on the hydration shell and intramolecular interactions.J Biol Inorg Chem. 2013 Feb;18(2):211-221. doi: 10.1007/s00775-012-0966-0. Epub 2013 Jan 16. J Biol Inorg Chem. 2013. PMID: 23322168
-
To Keep or Not to Keep? The Question of Crystallographic Waters for Enzyme Simulations in Organic Solvent.Mol Simul. 2016;42(12):1001-1013. doi: 10.1080/08927022.2016.1139108. Epub 2016 Mar 22. Mol Simul. 2016. PMID: 27403032 Free PMC article.
-
Modeling of solvent-dependent conformational transitions in Burkholderia cepacia lipase.BMC Struct Biol. 2009 May 28;9:38. doi: 10.1186/1472-6807-9-38. BMC Struct Biol. 2009. PMID: 19476626 Free PMC article.
References
-
- Allen, K. N., C. R. Bellamacina, X. Ding, C. J. Jeffery, C. Mattos, G. A. Petsko, and D. Ringe. 1996. An experimental approach to mapping the binding surfaces of crystalline proteins. J. Phys. Chem. 100:2605–2611.
-
- Baptista, A. M., and C. M. Soares. 2001. Some theoretical and computational aspects of the inclusion of proton isomerism in the protonation equilibrium of proteins. J. Phys. Chem. B. 105:293–309.
-
- Baptista, A. M., V. H. Teixeira, and C. M. Soares. 2002. Constant-pH molecular dynamics using stochastic titration. J. Chem. Phys. 17:4184–4200.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
