

doi: 10.1016/S0006-3495(03)74998-4.
Computer simulations of membrane protein folding: structure and dynamics
Affiliations
- PMID: 12609892
- PMCID: PMC1302759
- DOI: 10.1016/S0006-3495(03)74998-4
Item in Clipboard
Computer simulations of membrane protein folding: structure and dynamics
Biophys J.
2003 Mar.
Abstract
A lattice model of membrane proteins with a composite energy function is proposed to study their folding dynamics and native structures using Monte Carlo simulations. This model successfully predicts the seven helix bundle structure of sensory rhodopsin I by practicing a three-stage folding. Folding dynamics of a transmembrane segment into a helix is further investigated by varying the cooperativity in the formation of alpha helices for both random folding and assisted folding. The chain length dependence of the folding time of a hydrophobic segment to a helical state is studied for both free and anchored chains. An unusual length dependence in the folding time of anchored chains is observed.
Figures
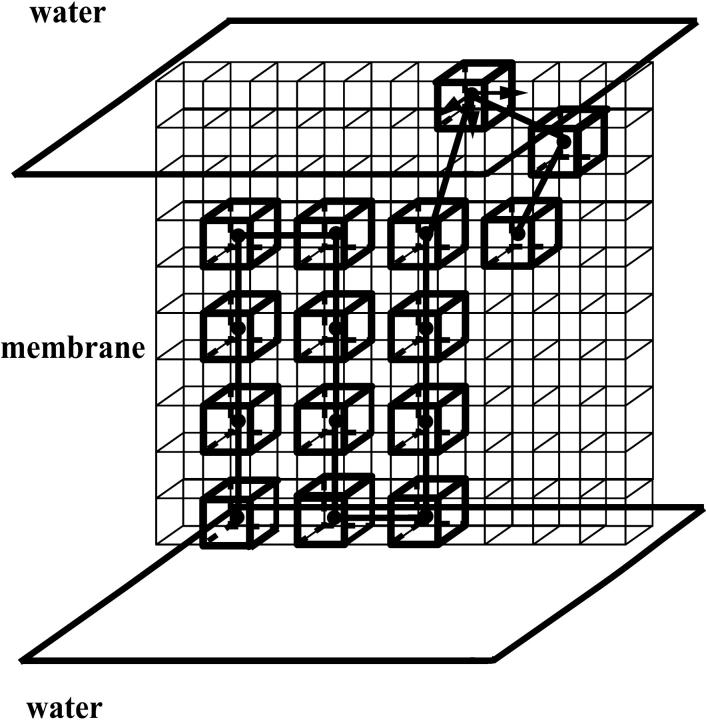
One protein chain of 15 residues confined in a membrane is shown. The membrane phase separates two water phases. All residues are shown in one plane for convenience, although the simulations are done in three dimensions.
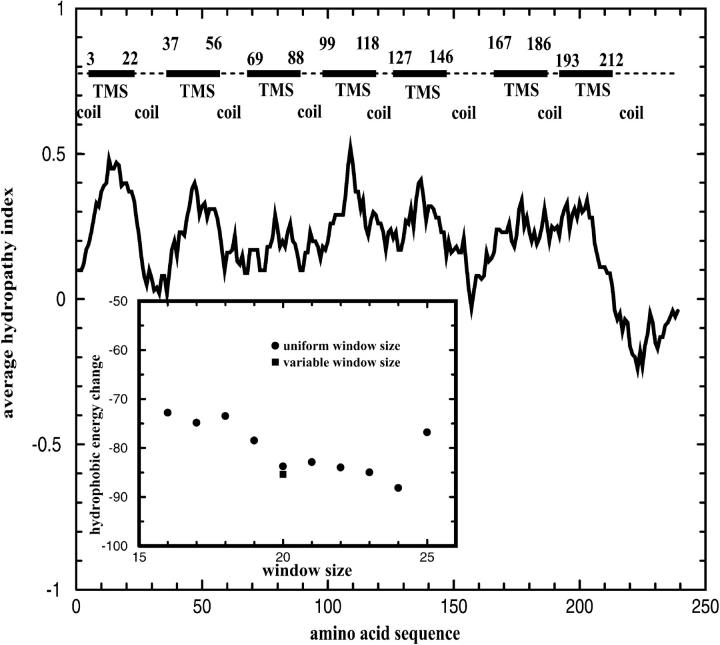
The average hydropathy index of SRI for a window of 20 amino acids. Seven transmembrane segments (TMSs) are predicted for SRI from optimizing the hydropathical interaction. The schematic representation of SRI above the hydropathy profile shows seven TMSs (filled rectangles) and eight coils (dash lines). The inset shows the hydrophobic energy reduction for various window sizes. Filled circles are results from using uniform window size ranging from 16 to 25. The filled square is a further minimization of the hydrophobic energy by varying the length of each transmembrane segment obtained from using window size 20.
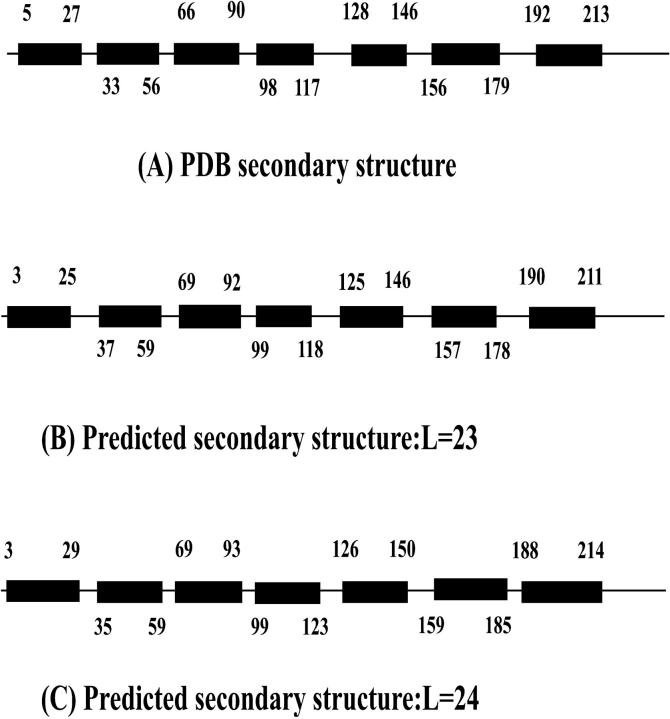
A comparison of the PDB secondary structure (A) and our predictions of SRI for L = 23 (B) and L = 24 (C). Each helix is represented by a filled rectangle. Those numbers along the chain label the corresponding amino acids at both ends of transmembrane helices. The parameters used are e1 = 0.3, e2 = 0, e3 = 0.3, e4 = 1.5, P = 0.1.
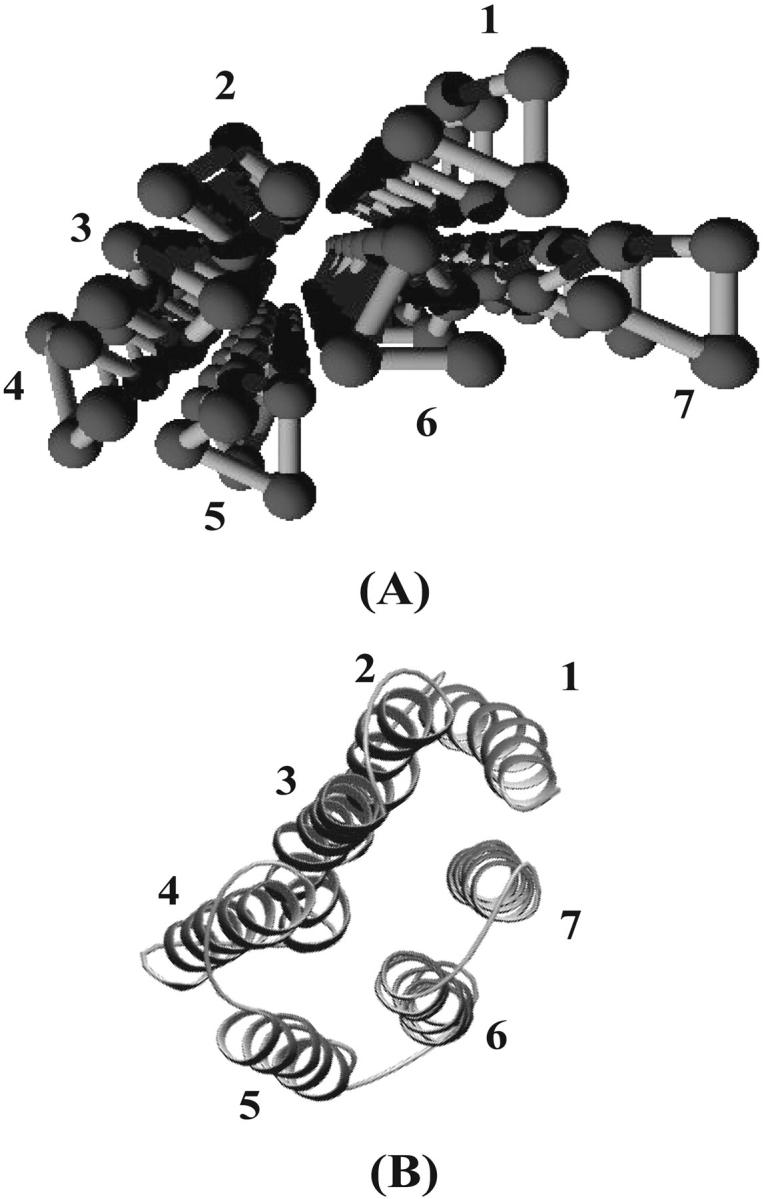
A comparison of the PDB tertiary structure (A) and our prediction (B) of SRI. Seven helices are labeled according to their position along the sequence. Flexible regions of SRI are not shown in (A). The parameters used are e1 = 0.3, e2 = 0.3, e3 = 0.3, e4 = 1.5, P = 0.1.
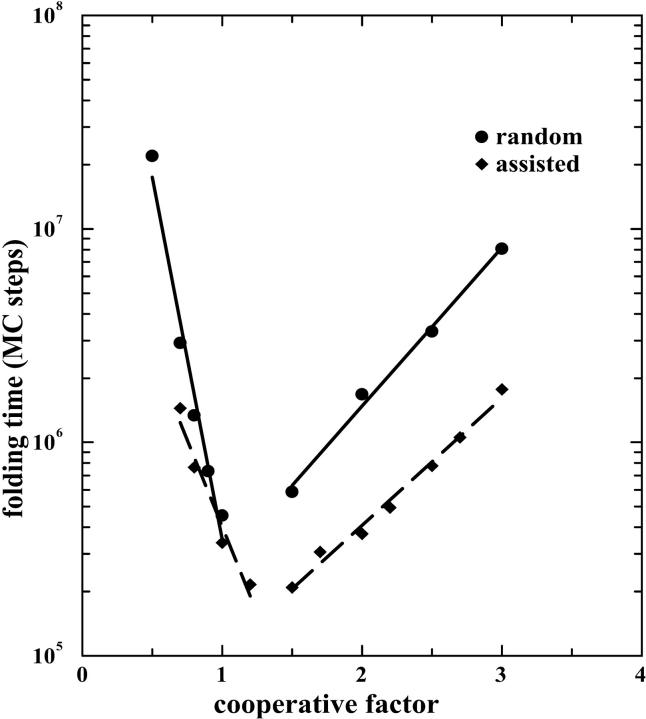
The dependence of folding time of a transmembrane helix on cooperative factor for both random folding and assisted folding. The parameters used are e1 = 0.3, e2 = 0, e3 = 0.3, e4 = 1.5, P = 0.1, T = 0.31, and L = 24.
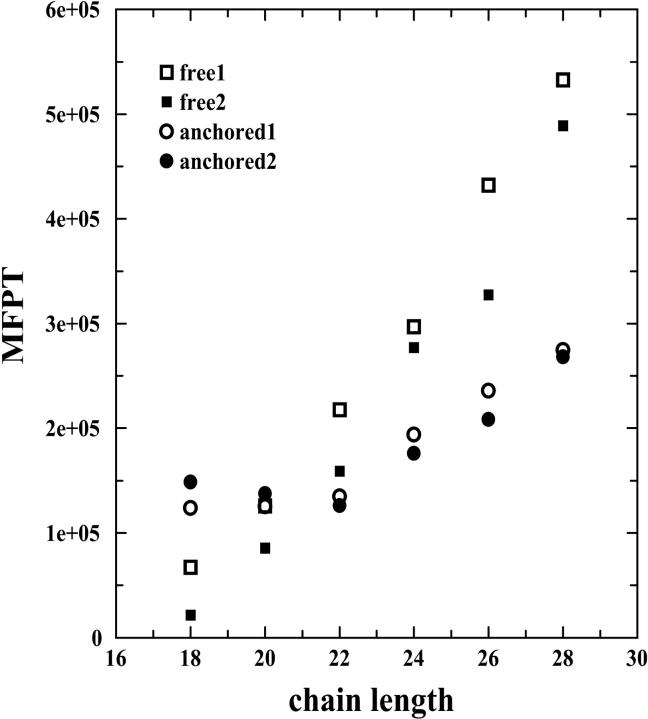
The dependence of the MFPT of a transmembrane helix on chain length for both free (open squares: rotating kinetics 1; filled squares: rotating kinetics 2) and anchored chains (open circles: rotating kinetics 1; filled circles: rotating kinetics 2). The parameters used are e1 = 0.3, e2 = 0, e3 = 0.3, e4 = 1.5, P = 0.1, T = 0.31, and L = 24.
Similar articles
-
Deciphering the folding kinetics of transmembrane helical proteins.Proc Natl Acad Sci U S A. 2000 Dec 19;97(26):14229-34. doi: 10.1073/pnas.97.26.14229. Proc Natl Acad Sci U S A. 2000. PMID: 11121029 Free PMC article.
-
Entropy reduction effect imposed by hydrogen bond formation on protein folding cooperativity: evidence from a hydrophobic minimalist model.Phys Rev E Stat Nonlin Soft Matter Phys. 2005 Nov;72(5 Pt 1):051903. doi: 10.1103/PhysRevE.72.051903. Epub 2005 Nov 1. Phys Rev E Stat Nonlin Soft Matter Phys. 2005. PMID: 16383641
-
Comparison of stability predictions and simulated unfolding of rhodopsin structures.Photochem Photobiol. 2007 Mar-Apr;83(2):351-62. doi: 10.1562/2006-06-20-RA-942. Photochem Photobiol. 2007. PMID: 17576347
-
A successful change of circumstance: a transition state for membrane protein folding.Curr Opin Struct Biol. 2012 Aug;22(4):469-75. doi: 10.1016/j.sbi.2012.03.008. Epub 2012 Apr 2. Curr Opin Struct Biol. 2012. PMID: 22475521 Review.
-
Dynamic Monte Carlo simulations of a new lattice model of globular protein folding, structure and dynamics.J Mol Biol. 1991 Sep 20;221(2):499-531. doi: 10.1016/0022-2836(91)80070-b. J Mol Biol. 1991. PMID: 1920430 Review.
Cited by
-
Conformational temperature-dependent behavior of a histone H2AX: a coarse-grained Monte Carlo approach via knowledge-based interaction potentials.PLoS One. 2012;7(3):e32075. doi: 10.1371/journal.pone.0032075. Epub 2012 Mar 19. PLoS One. 2012. PMID: 22442661 Free PMC article.
-
Contact-induced structure transformation in transmembrane prion propagation.Biophys J. 2007 Apr 15;92(8):2704-10. doi: 10.1529/biophysj.106.098335. Epub 2007 Jan 26. Biophys J. 2007. PMID: 17259269 Free PMC article.
-
Computational prediction of kink properties of helices in membrane proteins.J Comput Aided Mol Des. 2014 Feb;28(2):99-109. doi: 10.1007/s10822-014-9734-2. Epub 2014 Feb 21. J Comput Aided Mol Des. 2014. PMID: 24557854
-
Viral channel forming proteins--How to assemble and depolarize lipid membranes in silico.Biochim Biophys Acta. 2016 Jul;1858(7 Pt B):1710-21. doi: 10.1016/j.bbamem.2016.01.018. Epub 2016 Jan 22. Biochim Biophys Acta. 2016. PMID: 26806161 Free PMC article. Review.
-
Replica exchange Monte-Carlo simulations of helix bundle membrane proteins: rotational parameters of helices.J Comput Aided Mol Des. 2012 Mar;26(3):363-74. doi: 10.1007/s10822-012-9562-1. Epub 2012 Mar 31. J Comput Aided Mol Des. 2012. PMID: 22466784
References
-
- Arumugam, S., S. Pascal, C. L. North, W. Hu, K. C. Lee, M. Cotten, R. R. Ketchem, F. Xu, M. Brenneman, F. Kovacs, F. Tian, A. Wang, S. Huo, and T. A. Cross. 1996. Conformational trapping in a membrane environment: a regulatory mechanism for protein activity? Proc. Natl. Acad. Sci. U.S.A. 93:5872–5876. - PMC - PubMed
-
- Bryngelson, J. D., and P. G. Wolynes. 1989. Intermediates and barrier crossing in a random energy model (with applications to protein folding). J. Phys. Chem. 93:6902–6915.
-
- Cantor, C. R., and P. R. Schimmel. 1980. Biophysical Chemistry. W. H. Freeman Company, San Francisco. 862–863.
-
- Carmesin, I., and K. Kremer. 1988. The bond fluctuation method: a new effective algorithm for the dynamics of polymers in all spatial dimensions. Macromolecules. 21:2819–2823.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
