

Stability and Cu(II) binding of prion protein variants related to inherited human prion diseases
- PMID: 12609901
- PMCID: PMC1302768
- DOI: 10.1016/S0006-3495(03)75007-3
Stability and Cu(II) binding of prion protein variants related to inherited human prion diseases
Abstract
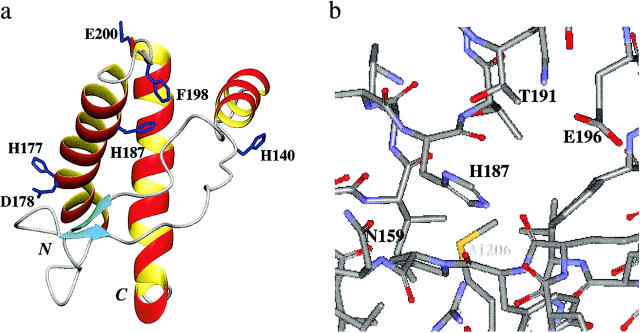
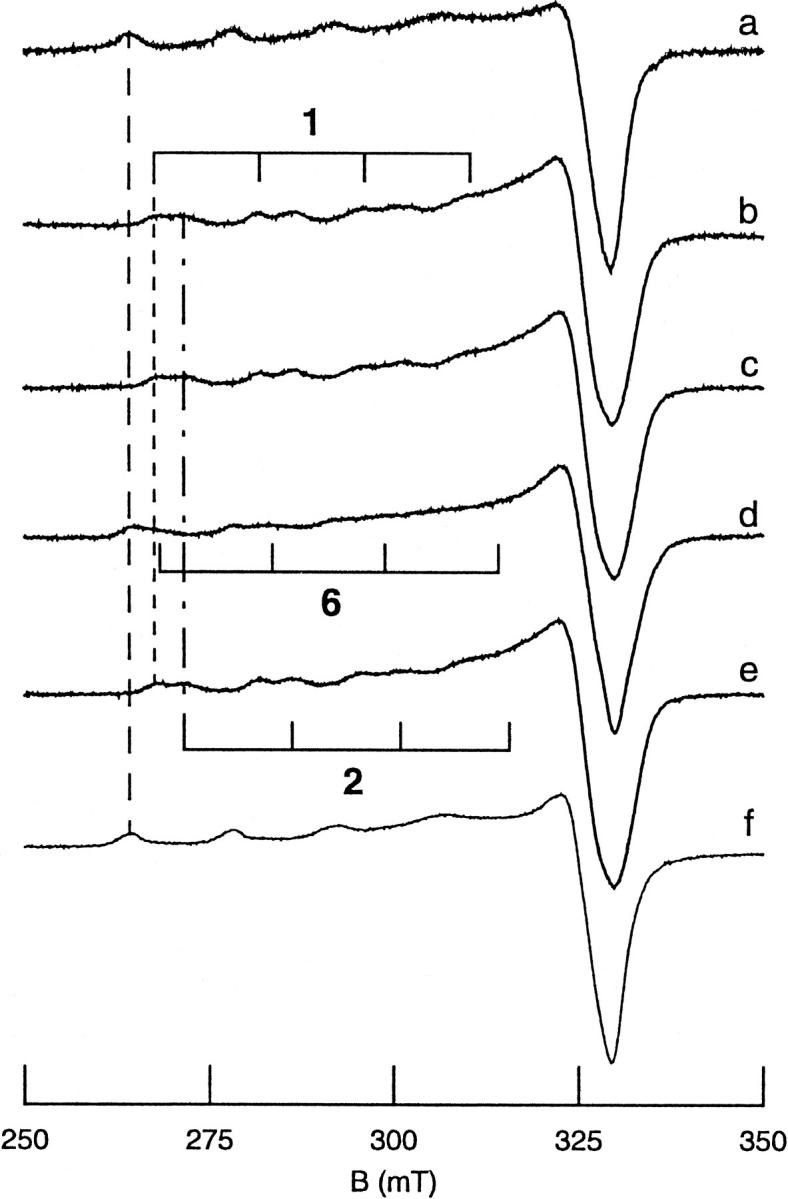
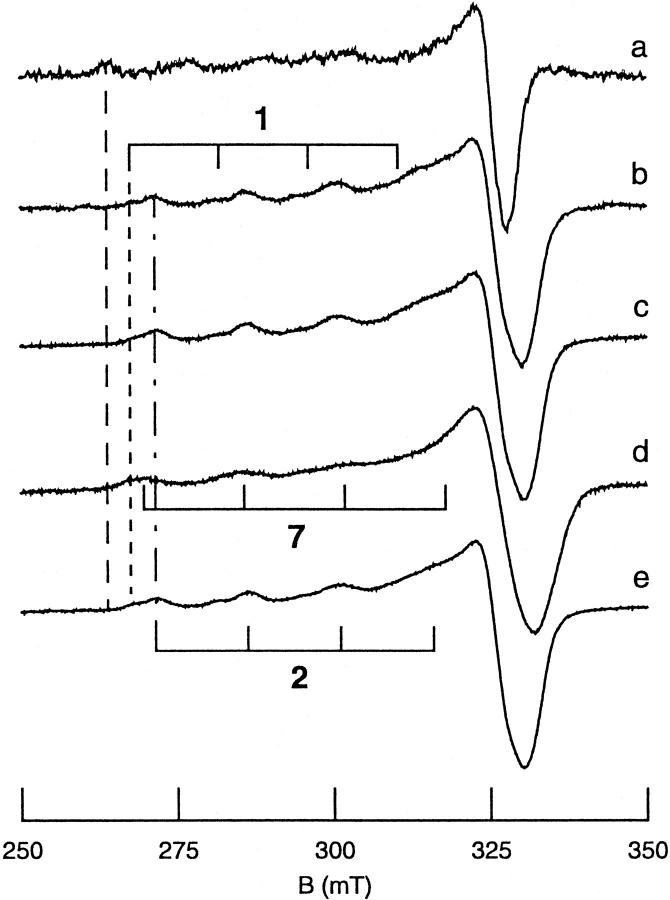
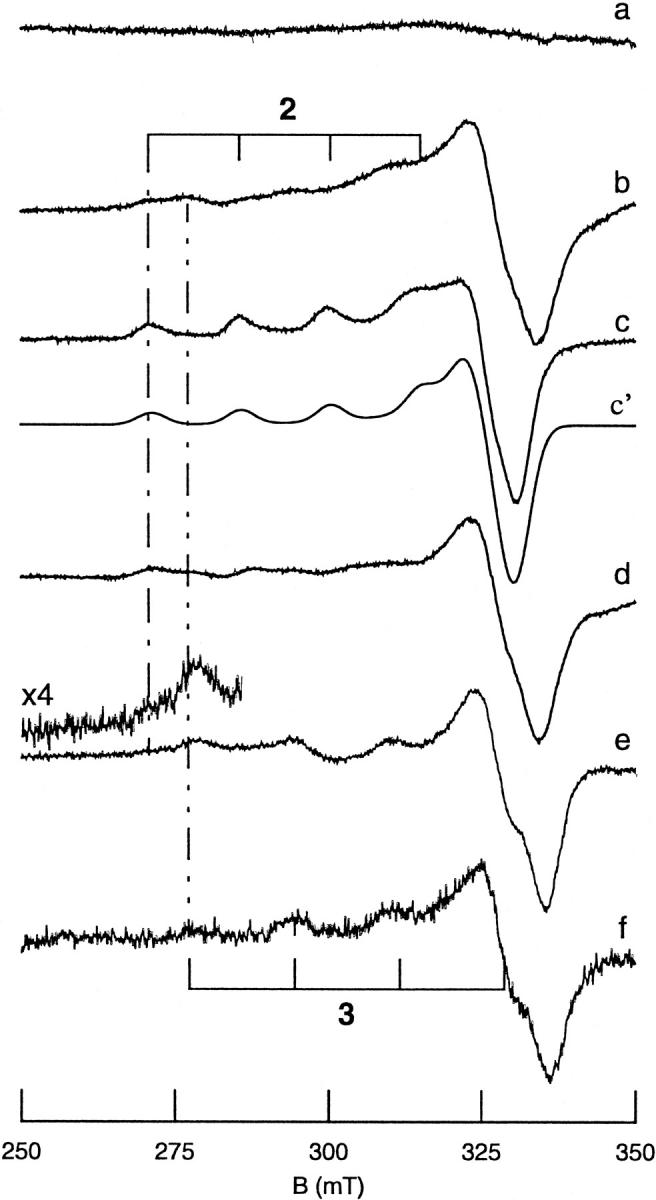
All inherited forms of human prion diseases are linked with mutations in the prion protein (PrP) gene. Here we have investigated the stability and Cu(II) binding properties of three recombinant variants of murine full-length PrP(23-231)-containing destabilizing point mutations that are associated with human Gerstmann-Sträussler-Scheinker disease (F198S), Creutzfeld-Jakob disease (E200K), and fatal familial insomnia (D178N) by electron paramagnetic resonance and circular dichroism spectroscopy. Furthermore, we analyzed the variants H140S, H177S, and H187S of the isolated C-terminal domain of murine PrP, mPrP(121-231), to test a role of the histidine residues in Cu(II) binding. The F198S and E200K variants of PrP(23-231) differed in Cu(II) binding from the wild-type mPrP(23-231). However, circular dichroism spectroscopy indicated that the variants and the wild type did not undergo conformational changes in the presence of Cu(II). The D178N variant showed a high tendency to aggregate at pH 7.4 both with and without Cu(II). At lower pH values, it showed the same Cu(II) binding behavior as the wild type. The analysis allowed for a better location of the Cu(II) binding sites in the C-terminal part of the protein. Our present data indicate that hereditary forms of prion diseases cannot be rationalized on the basis of altered Cu(II) binding or mutation-induced protein destabilization alone.
Figures
References
-
- Aronoff-Spencer, E., C. S. Burns, N. I. Avdievich, G. J. Gerfen, J. Peisach, W. E. Antholine, H. L. Ball, F. E. Cohen, S. B. Prusiner, and G. L. Millhauser. 2000. Identification of the Cu2+ binding sites in the N-terminal domain of the prion protein by EPR and CD spectroscopy. Biochemistry 39:13760–13771. - PMC - PubMed
-
- Bell, J. E. and J. W. Ironside. 1993. Neuropathology of spongiform encephalopathies in humans. Br. Med. Bull. 49:738–777. - PubMed
-
- Brown, D. R., K. Qin, J. W. Herms, A. Madlung, J. Manson, R. Strome, P. E. Fraser, T. Kruck, A. von Bohlen, W. Schulz-Schaeffer, A. Giese, D. Westaway, and H. Kretzschmar. 1997. The cellular prion protein binds copper in vivo. Nature 390:684–687. - PubMed
-
- Burns, C. S., E. Aronoff-Spencer, C. M. Dunham, P. Lario, N. I. Avdievich, W. E. Antholine, M. M. Olmstead, A. Vrielink, G. J. Gerfen, J. Peisach, W. G. Scott, and G. L. Millhauser. 2002. Molecular features of the copper-binding sites in the octarepeat domain of the prion protein. Biochemistry 41:3991–4001. - PMC - PubMed
-
- Butefisch, C. M., P. Gambetti, L. Cervenakova, K. Y. Park, M. Hallett, and L. G. Goldfarb. 2000. Inherited prion ecnephalopathy associated with the novel PRNP H187R mutation—a clinical study. Neurology 55:517–522. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
