

Myeloperoxidase and plasminogen activator inhibitor 1 play a central role in ventricular remodeling after myocardial infarction
- PMID: 12615902
- PMCID: PMC2193831
- DOI: 10.1084/jem.20021426
Myeloperoxidase and plasminogen activator inhibitor 1 play a central role in ventricular remodeling after myocardial infarction
Abstract
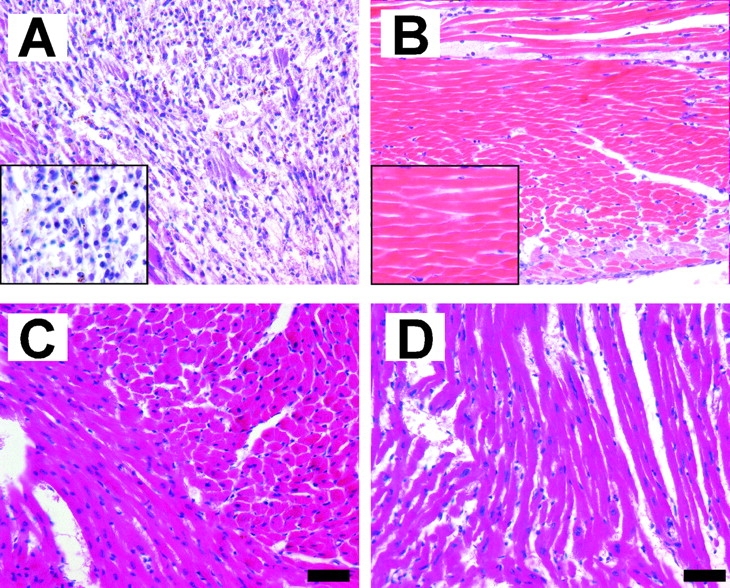
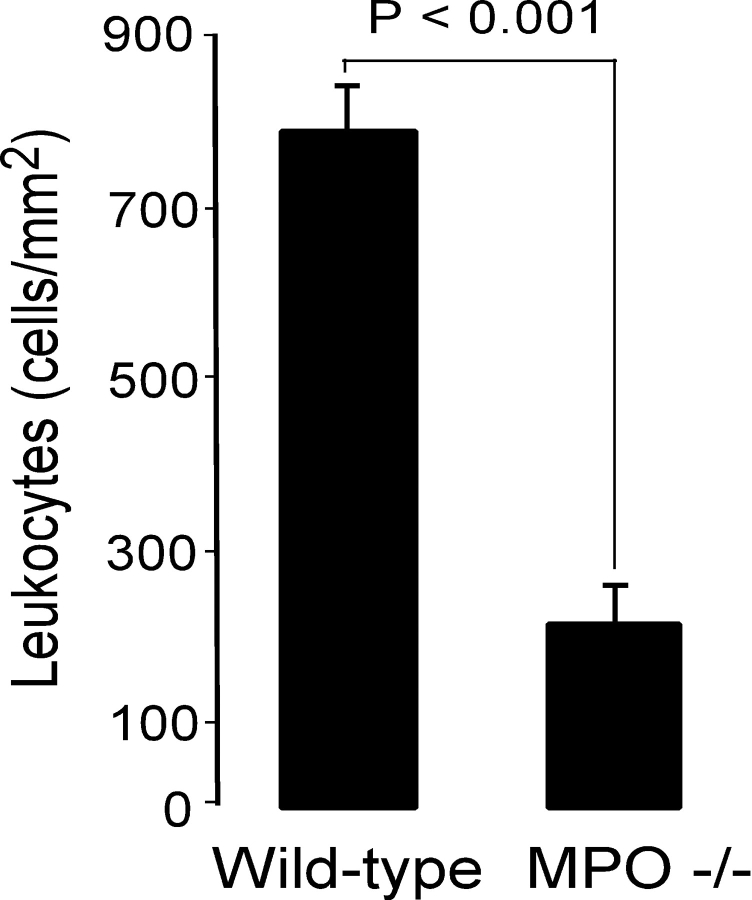
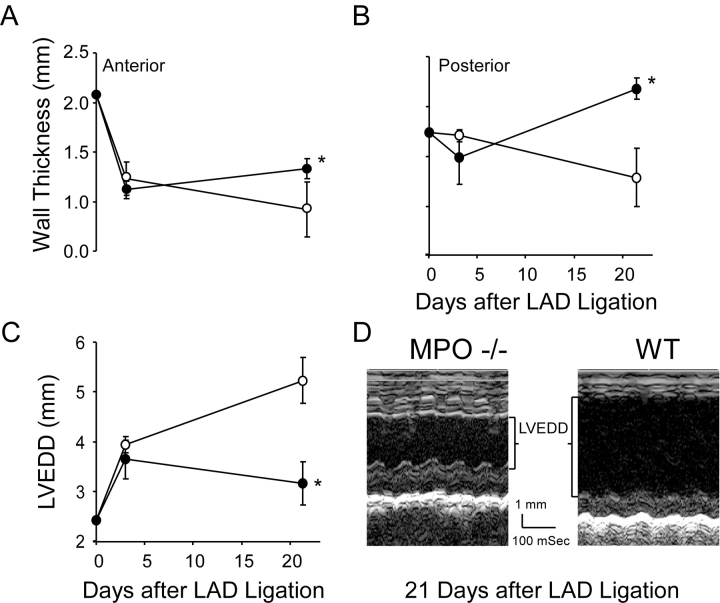
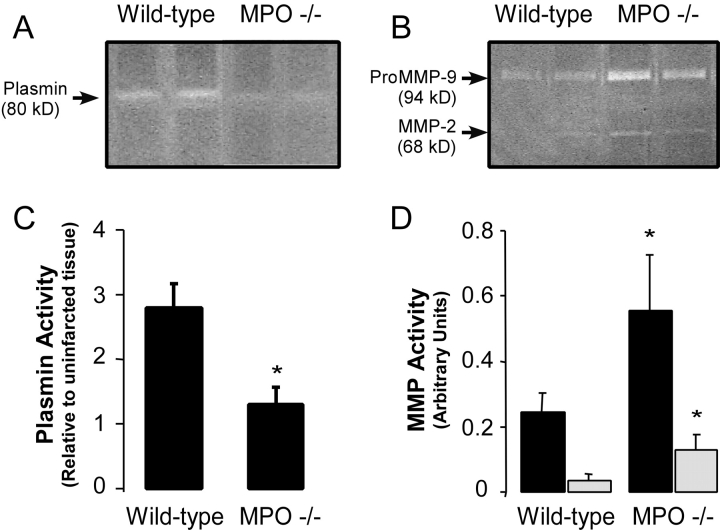
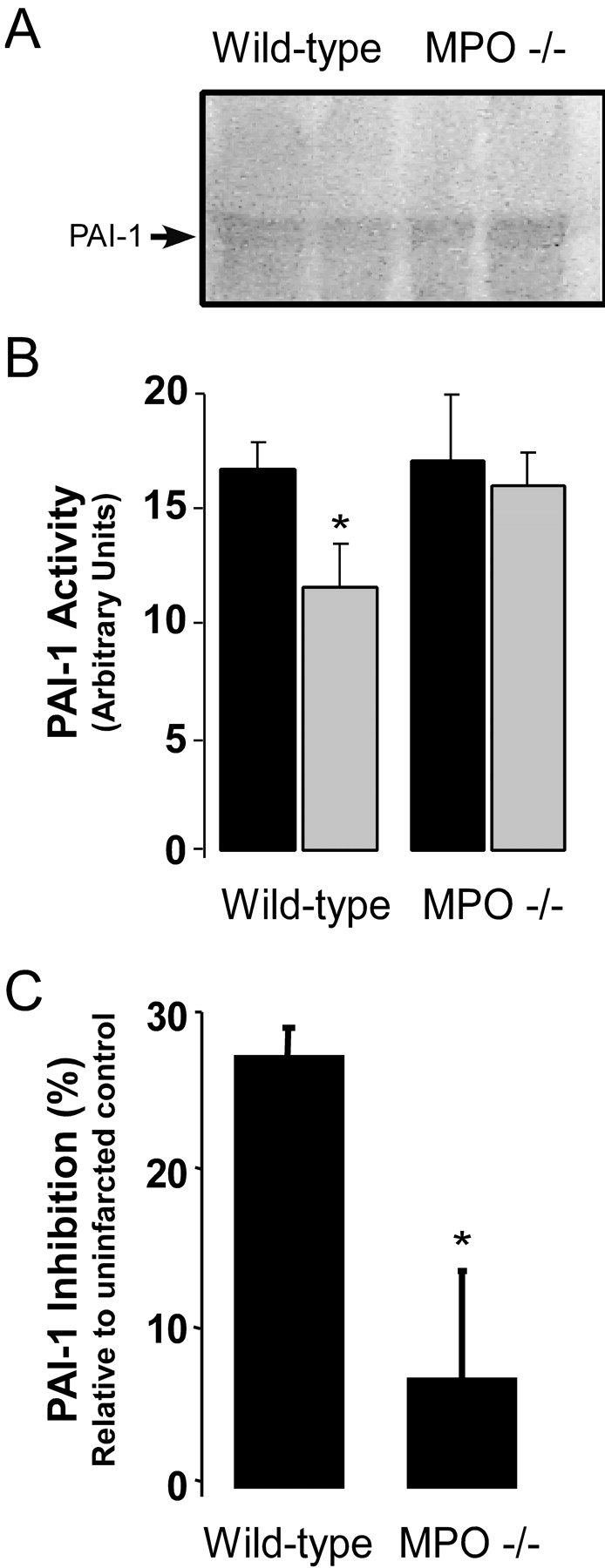
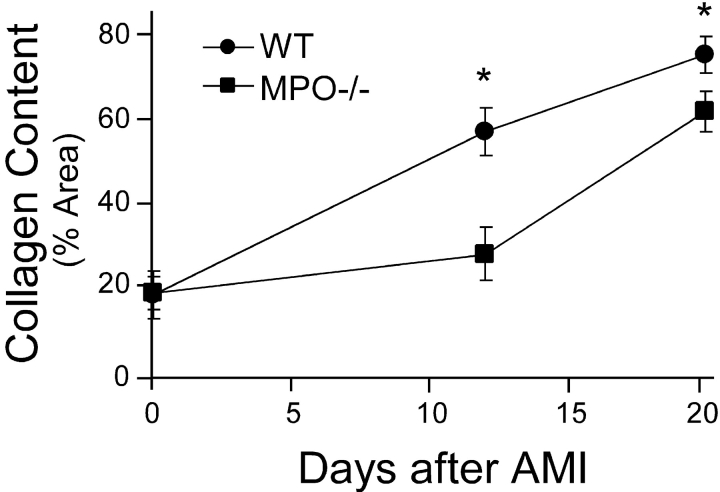
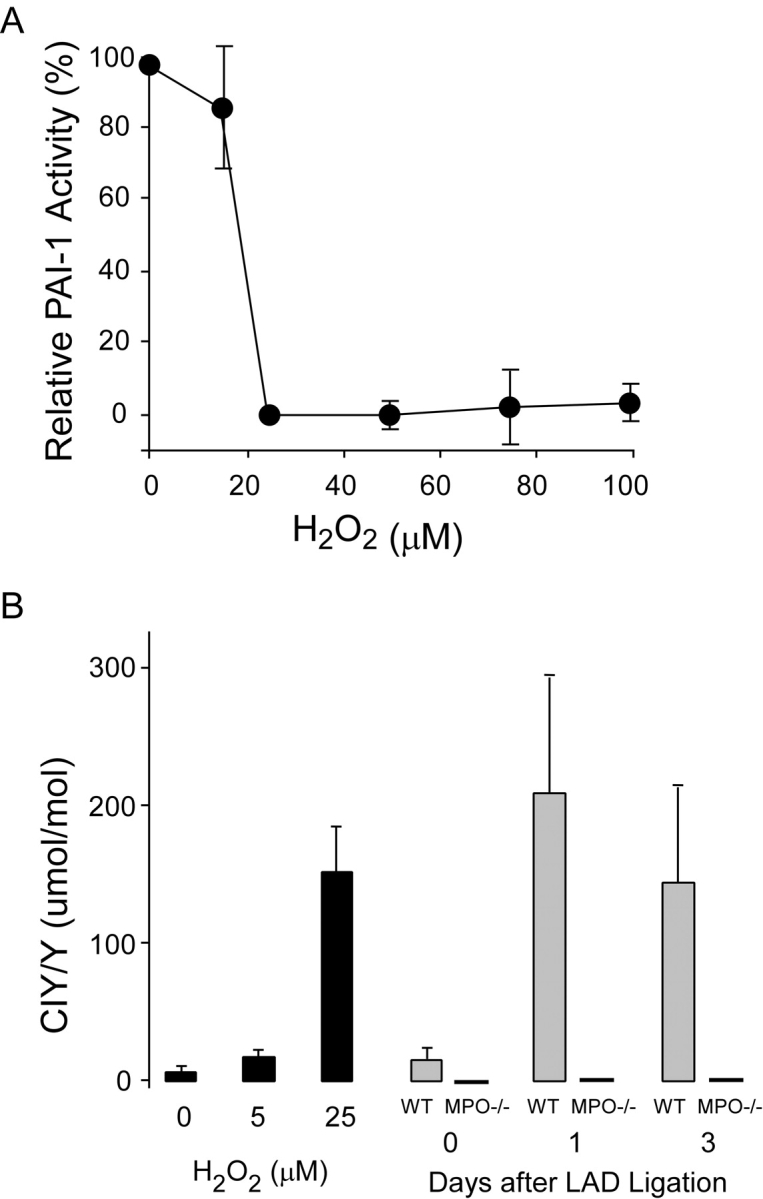
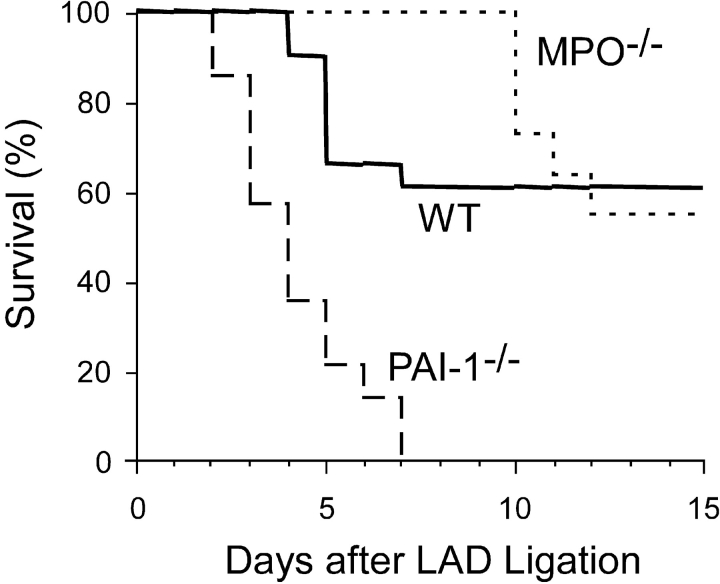
Left ventricular (LV) remodeling after myocardial infarction (MI) results in LV dilation, a major cause of congestive heart failure and sudden cardiac death. Ischemic injury and the ensuing inflammatory response participate in LV remodeling, leading to myocardial rupture and LV dilation. Myeloperoxidase (MPO), which accumulates in the infarct zone, is released from neutrophils and monocytes leading to the formation of reactive chlorinating species capable of oxidizing proteins and altering biological function. We studied acute myocardial infarction (AMI) in a chronic coronary artery ligation model in MPO null mice (MPO(-/-)). MPO(-/-) demonstrated decreased leukocyte infiltration, significant reduction in LV dilation, and marked preservation of LV function. The mechanism appears to be due to decreased oxidative inactivation of plasminogen activator inhibitor 1 (PAI-1) in the MPO(-/-), leading to decreased tissue plasmin activity. MPO and PAI-1 are shown to have a critical role in the LV response immediately after MI, as demonstrated by markedly delayed myocardial rupture in the MPO(-/-) and accelerated rupture in the PAI-1(-/-). These data offer a mechanistic link between inflammation and LV remodeling by demonstrating a heretofore unrecognized role for MPO and PAI-1 in orchestrating the myocardial response to AMI.
Figures
References
-
- St. John, S.M., M.A. Pfeffer, L. Moye, T. Plappert, J.L. Rouleau, G. Lamas, J. Rouleau, J.O. Parker, M.O. Arnold, B. Sussex, et al. 1997. Cardiovascular death and left ventricular remodeling two years after myocardial infarction: baseline predictors and impact of long-term use of captopril: information from the Survival and Ventricular Enlargement (SAVE) trial. Circulation. 96:3294–3299. - PubMed
-
- Groenning, B.A., J.C. Nilsson, L. Sondergaard, T. Fritz-Hansen, H.B. Larsson, and P.R. Hildebrandt. 2000. Antiremodeling effects on the left ventricle during beta-blockade with metoprolol in the treatment of chronic heart failure. J. Am. Coll. Cardiol. 36:2072–2080. - PubMed
-
- Heymans, S., A. Luttun, D. Nuyens, G. Theilmeier, E. Creemers, L. Moons, G.D. Dyspersin, J.P. Cleutjens, M. Shipley, A. Angellilo, et al. 1999. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat. Med. 5:1135–1142. - PubMed
-
- Ducharme, A., S. Frantz, M. Aikawa, E. Rabkin, M. Lindsey, L.E. Rohde, F.J. Schoen, R.A. Kelly, Z. Werb, P. Libby, et al. 2000. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J. Clin. Invest. 106:55–62. - PMC - PubMed
-
- Hazen, S.L., J.R. Crowley, D.M. Mueller, and J.W. Heinecke. 1997. Mass spectrometric quantification of 3-chlorotyrosine in human tissues with attomole sensitivity: a sensitive and specific marker for myeloperoxidase-catalyzed chlorination at sites of inflammation. Free Radic. Biol. Med. 23:909–916. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
