

Segmental duplications in euchromatic regions of human chromosome 5: a source of evolutionary instability and transcriptional innovation
- PMID: 12618367
- PMCID: PMC430257
- DOI: 10.1101/gr.490303
Segmental duplications in euchromatic regions of human chromosome 5: a source of evolutionary instability and transcriptional innovation
Abstract
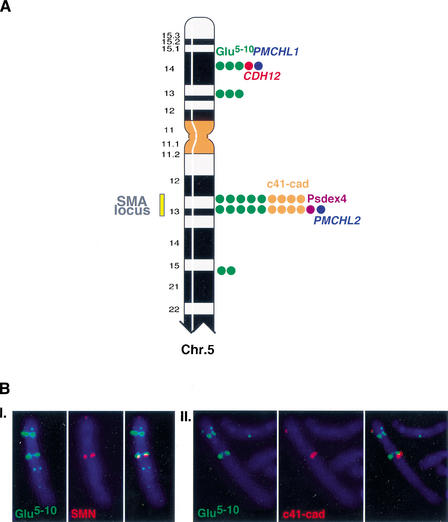
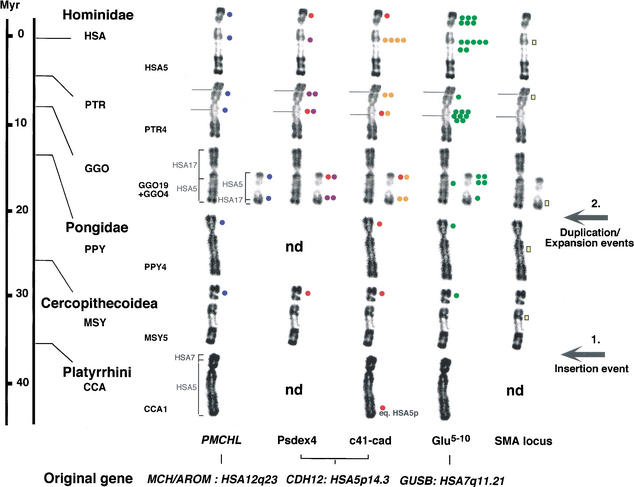
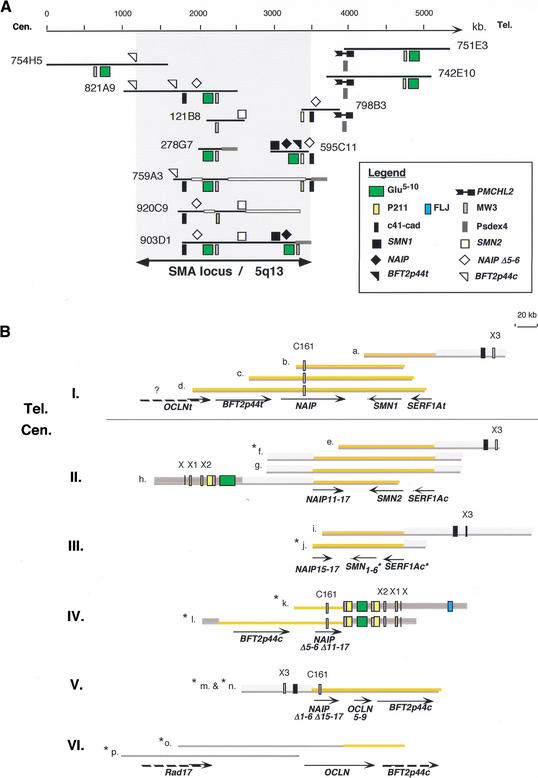
Recent analyses of the structure of pericentromeric and subtelomeric regions have revealed that these particular regions of human chromosomes are often composed of blocks of duplicated genomic segments that have been associated with rapid evolutionary turnover among the genomes of closely related primates. In the present study, we show that euchromatic regions of human chromosome 5-5p14, 5p13, 5q13, 5q15-5q21-also display such an accumulation of segmental duplications. The structure, organization and evolution of those primate-specific sequences were studied in detail by combining in silico and comparative FISH analyses on human, chimpanzee, gorilla, orangutang, macaca, and capuchin chromosomes. Our results lend support to a two-step model of transposition duplication in the euchromatic regions, with a founder insertional event at the time of divergence between Platyrrhini and Catarrhini (25-35 million years ago) and an apparent burst of inter- and intrachromosomal duplications in the Hominidae lineage. Furthermore, phylogenetic analysis suggests that the chronology and, likely, molecular mechanisms, differ regarding the region of primary insertion-euchromatic versus pericentromeric regions. Lastly, we show that as their counterparts located near the heterochromatic region, the euchromatic segmental duplications have consistently reshaped their region of insertion during primate evolution, creating putative mosaic genes, and they are obvious candidates for causing ectopic rearrangements that have contributed to evolutionary/genomic instability.
Figures



References
-
- Bailey J.A., Gu, Z., Clark, R.A., Reinert, K., Samonte, R.V., Schwartz, S., Adams, M.D., Myers, E.W., Li, P.W., and Eichler, E.E. 2002a. Recent segmental duplications in the human genome. Science 297: 1003-1007. - PubMed
-
- Brosius J. 1999a. RNAs from all categories generate retrosequences that may be exapted as novel genes or regulatory elements. Gene 238: 115-134. - PubMed
-
- ___, 1999b. Vertebrate genomes were forged by massive bombardments with retroelements and retrosequences. Genetica 107: 209-238. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources