

A highly efficient recombineering-based method for generating conditional knockout mutations
- PMID: 12618378
- PMCID: PMC430283
- DOI: 10.1101/gr.749203
A highly efficient recombineering-based method for generating conditional knockout mutations
Abstract
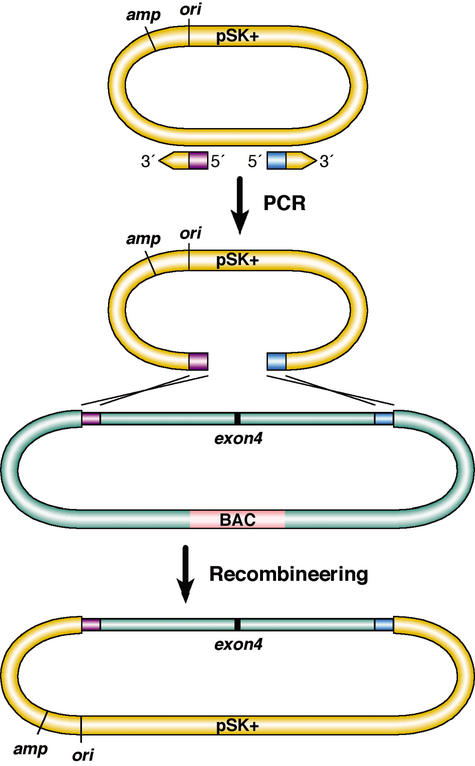
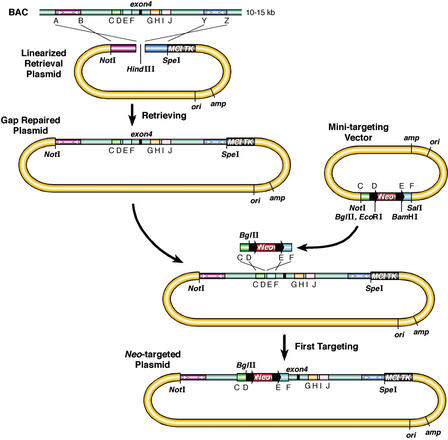
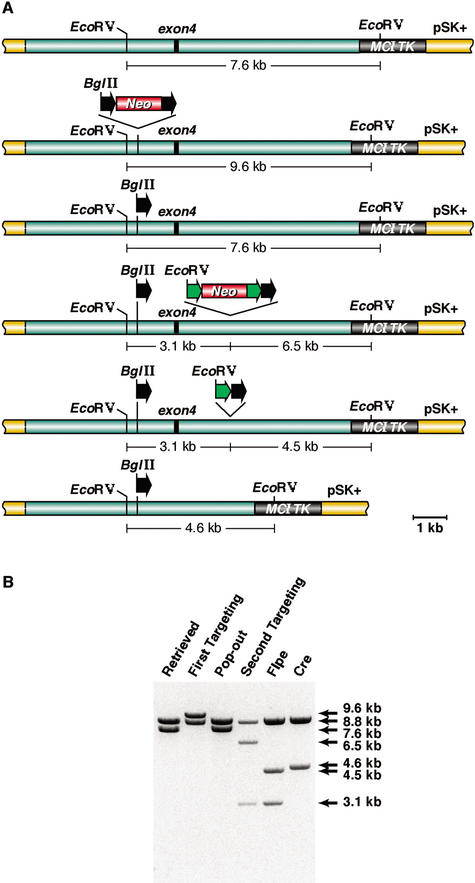
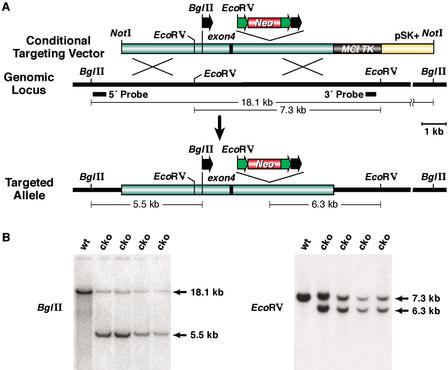
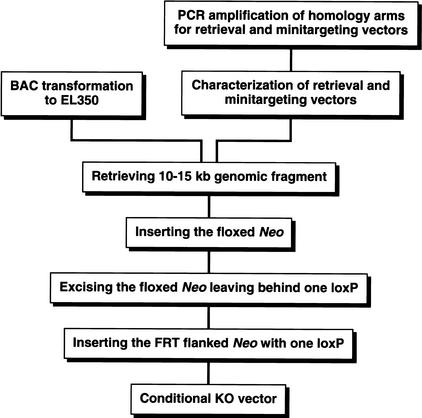
Phage-based Escherichia coli homologous recombination systems have recently been developed that now make it possible to subclone or modify DNA cloned into plasmids, BACs, or PACs without the need for restriction enzymes or DNA ligases. This new form of chromosome engineering, termed recombineering, has many different uses for functional genomic studies. Here we describe a new recombineering-based method for generating conditional mouse knockout (cko) mutations. This method uses homologous recombination mediated by the lambda phage Red proteins, to subclone DNA from BACs into high-copy plasmids by gap repair, and together with Cre or Flpe recombinases, to introduce loxP or FRT sites into the subcloned DNA. Unlike other methods that use short 45-55-bp regions of homology for recombineering, our method uses much longer regions of homology. We also make use of several new E. coli strains, in which the proteins required for recombination are expressed from a defective temperature-sensitive lambda prophage, and the Cre or Flpe recombinases from an arabinose-inducible promoter. We also describe two new Neo selection cassettes that work well in both E. coli and mouse ES cells. Our method is fast, efficient, and reliable and makes it possible to generate cko-targeting vectors in less than 2 wk. This method should also facilitate the generation of knock-in mutations and transgene constructs, as well as expedite the analysis of regulatory elements and functional domains in or near genes.
Figures
References
-
- Buchholz F., Angrand, P.O., and Stewart, A.F. 1998. Improved properties of FLP recombinase evolved by cycling mutagenesis. Nat. Biotechnol. 16: 657-662. - PubMed
-
- Copeland N.G., Jenkins, N.A., and Court, D.L. 2001. Mouse genomic technologies recombineering: A powerful new tool for mouse functional genomics. Nat. Rev. Genet. 2: 769-779. - PubMed
-
- Courcelle J. and Hanawalt, P.C. 1999. RecQ and RecJ process blocked replication forks prior to the resumption of replication in UV-irradiated Escherichia coli. Mol. Gen. Genet. 262: 543-551. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous