

SLAM: cross-species gene finding and alignment with a generalized pair hidden Markov model
- PMID: 12618381
- PMCID: PMC430255
- DOI: 10.1101/gr.424203
SLAM: cross-species gene finding and alignment with a generalized pair hidden Markov model
Abstract
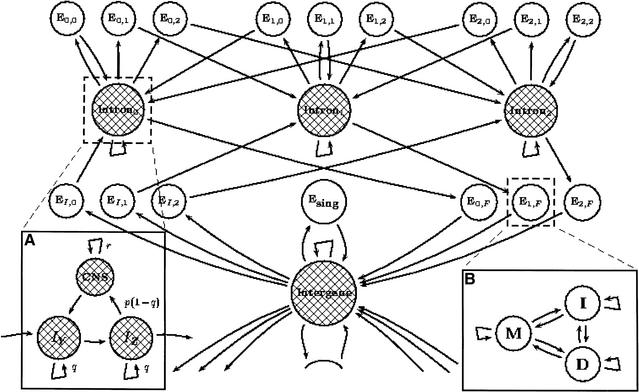
Comparative-based gene recognition is driven by the principle that conserved regions between related organisms are more likely than divergent regions to be coding. We describe a probabilistic framework for gene structure and alignment that can be used to simultaneously find both the gene structure and alignment of two syntenic genomic regions. A key feature of the method is the ability to enhance gene predictions by finding the best alignment between two syntenic sequences, while at the same time finding biologically meaningful alignments that preserve the correspondence between coding exons. Our probabilistic framework is the generalized pair hidden Markov model, a hybrid of (1). generalized hidden Markov models, which have been used previously for gene finding, and (2). pair hidden Markov models, which have applications to sequence alignment. We have built a gene finding and alignment program called SLAM, which aligns and identifies complete exon/intron structures of genes in two related but unannotated sequences of DNA. SLAM is able to reliably predict gene structures for any suitably related pair of organisms, most notably with fewer false-positive predictions compared to previous methods (examples are provided for Homo sapiens/Mus musculus and Plasmodium falciparum/Plasmodium vivax comparisons). Accuracy is obtained by distinguishing conserved noncoding sequence (CNS) from conserved coding sequence. CNS annotation is a novel feature of SLAM and may be useful for the annotation of UTRs, regulatory elements, and other noncoding features.
Figures

References
-
- Altschul S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215: 403-410. - PubMed
-
- Bafna V. and Huson, D.H. 2000. The conserved exon method for gene finding. ISMB-00: Proceedings of the Eight International Conference on Intelligent systems for Molecular Biology. 8: 3-12. - PubMed
-
- Bergman C.M. and Kreitman, M. 2001. Analysis of conserved noncoding DNA in Drosophila reveals similar constraints in intergenic and intronic sequences. Genet. Res. 11: 1335-1345. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical