

Large-scale identification of single-feature polymorphisms in complex genomes
- PMID: 12618383
- PMCID: PMC430246
- DOI: 10.1101/gr.541303
Large-scale identification of single-feature polymorphisms in complex genomes
Abstract
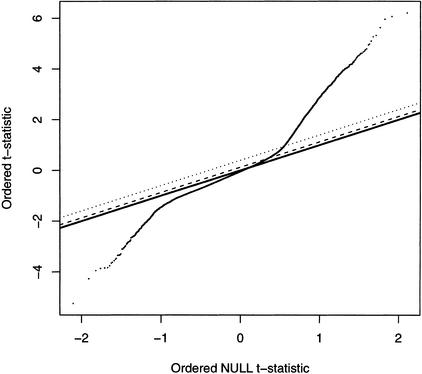
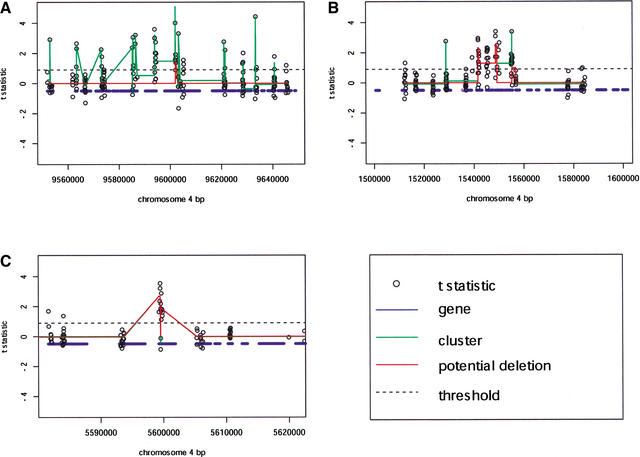
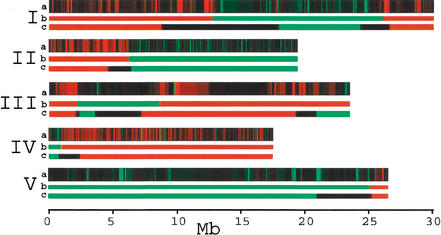
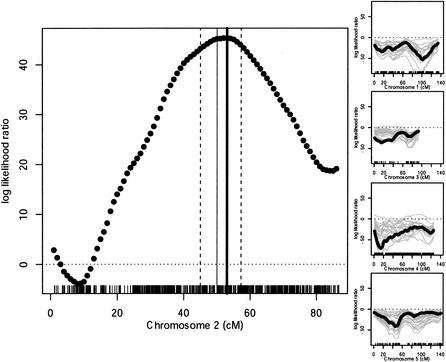
We have developed a high-throughput genotyping platform by hybridizing genomic DNA from Arabidopsis thaliana accessions to an RNA expression GeneChip (AtGenome1). Using newly developed analytical tools, a large number of single-feature polymorphisms (SFPs) were identified. A comparison of two accessions, the reference strain Columbia (Col) and the strain Landsberg erecta (Ler), identified nearly 4000 SFPs, which could be reliably scored at a 5% error rate. Ler sequence was used to confirm 117 of 121 SFPs and to determine the sensitivity of array hybridization. Features containing sequence repeats, as well as those from high copy genes, showed greater polymorphism rates. A linear clustering algorithm was developed to identify clusters of SFPs representing potential deletions in 111 genes at a 5% false discovery rate (FDR). Among the potential deletions were transposons, disease resistance genes, and genes involved in secondary metabolism. The applicability of this technique was demonstrated by genotyping a recombinant inbred line. Recombination break points could be clearly defined, and in one case delimited to an interval of 29 kb. We further demonstrate that array hybridization can be combined with bulk segregant analysis to quickly map mutations. The extension of these tools to organisms with complex genomes, such as Arabidopsis, will greatly increase our ability to map and clone quantitative trait loci (QTL).
Figures

References
-
- Bell C.J. and Ecker, J.R. 1994. Assignment of 30 microsatellite loci to the linkage map of Arabidopsis. Genomics 19: 137-144. - PubMed
-
- Brem R.B., Yvert, G., Clinton, R., and Kruglyak, L. 2002. Genetic dissection of transcriptional regulation in budding yeast. Science 296: 752-755. - PubMed
-
- Cho R.J., Mindrinos, M., Richards, D.R., Sapolsky, R.J., Anderson, M., Drenkard, E., Dewdney, J., Reuber, T.L., Stammers, M., Federspiel, N., et al. 1999. Genome-wide mapping with biallelic markers in Arabidopsis thaliana. Nat. Genet. 23: 203-207. - PubMed
-
- Doerge R.W. 2002. Mapping and analysis of quantitative trait loci in experimental populations. Nat. Rev. Genet. 3: 43-52. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical