

Toucan: deciphering the cis-regulatory logic of coregulated genes
- PMID: 12626717
- PMCID: PMC152870
- DOI: 10.1093/nar/gkg268
Toucan: deciphering the cis-regulatory logic of coregulated genes
Abstract
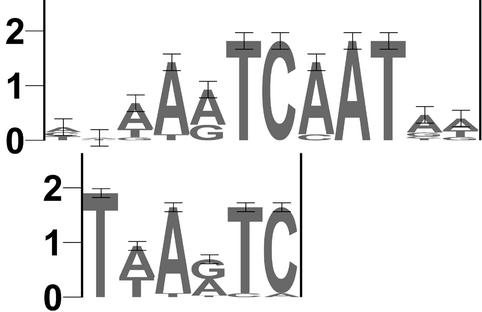
TOUCAN is a Java application for the rapid discovery of significant cis-regulatory elements from sets of coexpressed or coregulated genes. Biologists can automatically (i) retrieve genes and intergenic regions, (ii) identify putative regulatory regions, (iii) score sequences for known transcription factor binding sites, (iv) identify candidate motifs for unknown binding sites, and (v) detect those statistically over-represented sites that are characteristic for a gene set. Genes or intergenic regions are retrieved from Ensembl or EMBL, together with orthologs and supporting information. Orthologs are aligned and syntenic regions are selected as candidate regulatory regions. Putative sites for known transcription factors are detected using our MotifScanner, which scores position weight matrices using a probabilistic model. New motifs are detected using our MotifSampler based on Gibbs sampling. Binding sites characteristic for a gene set--and thus statistically over-represented with respect to a reference sequence set--are found using a binomial test. We have validated Toucan by analyzing muscle-specific genes, liver-specific genes and E2F target genes; we have easily detected many known binding sites within intergenic DNA and identified new biologically plausible sites for known and unknown transcription factors. Software available at http://www.esat.kuleuven.ac. be/ approximately dna/BioI/Software.html.
Figures
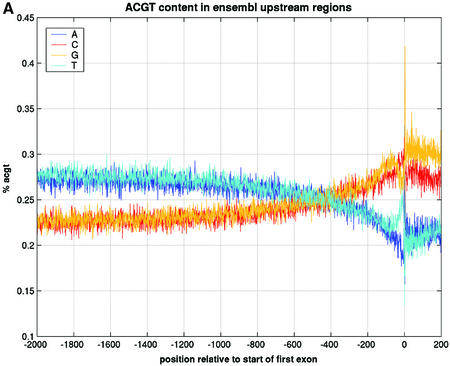
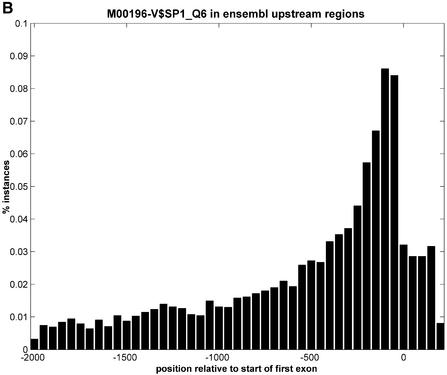
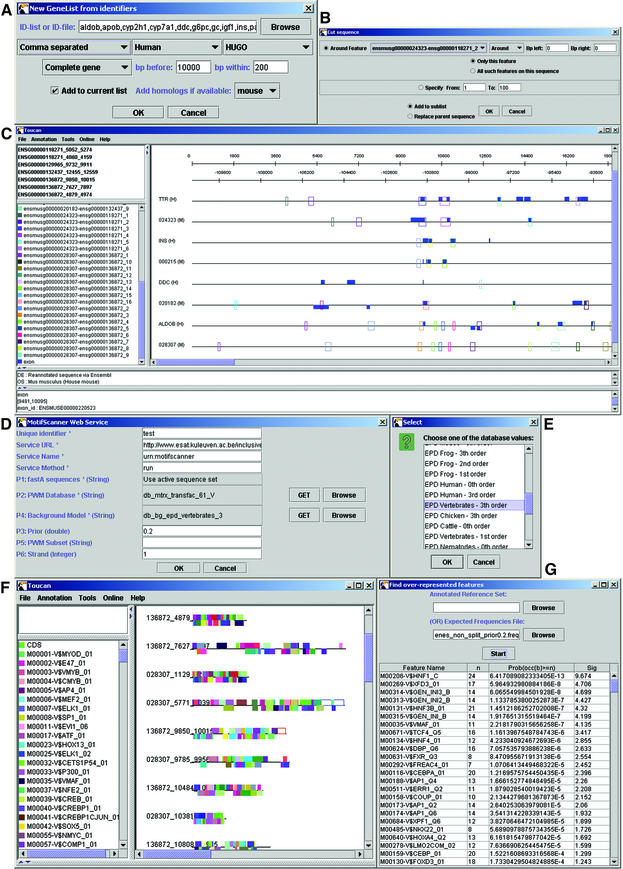
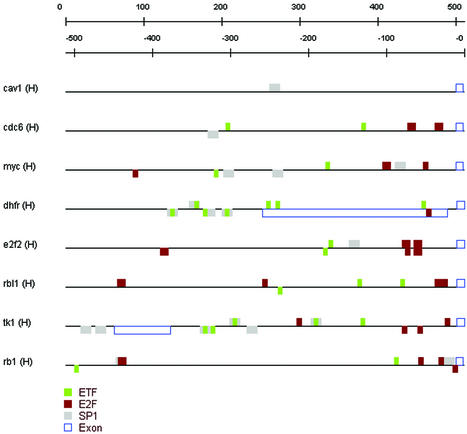
References
-
- Lemon B. and Tjian,R. (2000) Orchestrated response: a symphony of transcription factors for gene control. Genes Dev., 14, 2551–2569. - PubMed
-
- Davidson E.H. (2001) Genomic Regulatory Systems. Development and Evolution. Academic Press, San Diego, CA.
-
- Hughes J.D., Estep,P.W., Tavazoie,S. and Church,G.M. (2000) Computational identification of cis-regulatory elements associated with groups of functionally related genes in Saccharomyces cerevisiae. J. Mol. Biol., 296, 1205–1214. - PubMed
-
- Thijs G., Marchal,K., Lescot,M., Rombouts,S., De Moor,B., Rouze,P. and Moreau,Y. (2002) A Gibbs sampling method to detect overrepresented motifs in the upstream regions of coexpressed genes. J. Comput. Biol., 9, 447–464. - PubMed
-
- Berman B.P., Nibu,Y., Pfeiffer,B.D., Tomancak,P., Celniker,S.E., Levine,M., Rubin,G.M. and Eisen,M.B. (2002) Exploiting transcription factor binding site clustering to identify cis-regulatory modules involved in pattern formation in the Drosophila genome. Proc. Natl Acad. Sci. USA, 99, 757–762. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
