

Missense mutations in CRELD1 are associated with cardiac atrioventricular septal defects
- PMID: 12632326
- PMCID: PMC1180336
- DOI: 10.1086/374319
Missense mutations in CRELD1 are associated with cardiac atrioventricular septal defects
Abstract
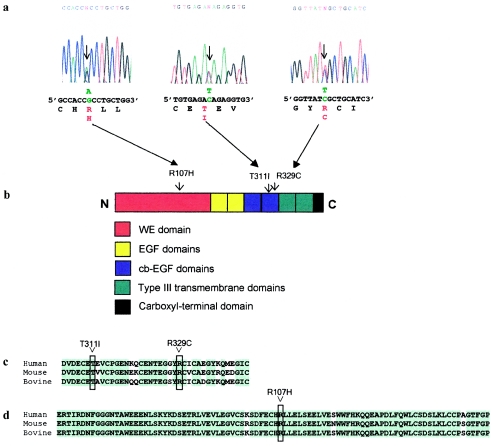
Atrioventricular septal defects (AVSD) are common cardiovascular malformations, occurring in 3.5/10,000 births. Although frequently associated with trisomy 21, autosomal dominant AVSD has also been described. Recently we identified and characterized the cell adhesion molecule CRELD1 (previously known as "cirrin") as a candidate gene for the AVSD2 locus mapping to chromosome 3p25. Analysis of the CRELD1 gene from individuals with non-trisomy 21-associated AVSD identified heterozygous missense mutations in nearly 6% of this population, including mutations in isolated AVSD and AVSD associated with heterotaxy syndrome. CRELD1 is the first human gene to be implicated in the pathogenesis of isolated AVSD and AVSD in the context of heterotaxy, which provides an important step in unraveling the pathogenesis of AVSD.
Figures


Similar articles
-
Molecular genetics of atrioventricular septal defects.Curr Opin Cardiol. 2004 May;19(3):205-10. doi: 10.1097/00001573-200405000-00003. Curr Opin Cardiol. 2004. PMID: 15096951 Review.
-
CRELD1 gene variants and atrioventricular septal defects in Down syndrome.Gene. 2018 Jan 30;641:180-185. doi: 10.1016/j.gene.2017.10.044. Epub 2017 Oct 18. Gene. 2018. PMID: 29054759
-
Analysis of CRELD1 as a candidate 3p25 atrioventicular septal defect locus (AVSD2).Clin Genet. 2005 Jun;67(6):526-8. doi: 10.1111/j.1399-0004.2005.00435.x. Clin Genet. 2005. PMID: 15857420 No abstract available.
-
[Potential role of CRELD1 gene in the pathogenesis of atrioventricular septal defect].Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2014 Jun;31(3):263-7. doi: 10.3760/cma.j.issn.1003-9406.2014.03.001. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2014. PMID: 24927998 Chinese.
-
[Molecular genetics of atrioventricular septal defects].Zhonghua Er Ke Za Zhi. 2005 May;43(5):390-2. Zhonghua Er Ke Za Zhi. 2005. PMID: 15924761 Review. Chinese. No abstract available.
Cited by
-
RTN3 inducing apoptosis is modulated by an adhesion protein CRELD1.Mol Cell Biochem. 2009 Nov;331(1-2):225-30. doi: 10.1007/s11010-009-0163-9. Epub 2009 Jun 12. Mol Cell Biochem. 2009. PMID: 19521671
-
Gender differences in the prevalence of congenital heart disease in Down's syndrome: a brief meta-analysis.BMC Med Genet. 2017 Oct 6;18(1):111. doi: 10.1186/s12881-017-0475-7. BMC Med Genet. 2017. PMID: 28985718 Free PMC article.
-
CRELD1 modulates homeostasis of the immune system in mice and humans.Nat Immunol. 2020 Dec;21(12):1517-1527. doi: 10.1038/s41590-020-00811-2. Epub 2020 Nov 9. Nat Immunol. 2020. PMID: 33169013
-
A new syndrome with noncompaction cardiomyopathy, bradycardia, pulmonary stenosis, atrial septal defect and heterotaxy with suggestive linkage to chromosome 6p.Hum Genet. 2008 Jan;122(6):595-603. doi: 10.1007/s00439-007-0436-x. Epub 2007 Oct 16. Hum Genet. 2008. PMID: 17938964
-
Genome-wide identification of mouse congenital heart disease loci.Hum Mol Genet. 2010 Aug 15;19(16):3105-13. doi: 10.1093/hmg/ddq211. Epub 2010 May 28. Hum Mol Genet. 2010. PMID: 20511334 Free PMC article.
References
Electronic-Database Information
-
- GenBank, http://www.ncbi.nlm.nih.gov/GenBank/ (for the previously published cDNA sequence for human CRELD1 [accession number AF452623])
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for Ivemark syndrome, autosomal heterotaxy syndrome, 3p− syndrome, Ellis-van Creveld syndrome, CHARGE syndrome, Kaufman-McKusick syndrome, AVSD1, AVSD2, and CRELD1)
References
-
- Amati F, Mari A, Mingarelli R, Gennarelli M, Digilio MC, Giannotti A, Marino B, Novelli G, Dallapiccola B (1995) Two pedigrees of autosomal dominant atrioventricular canal defect (AVCD): exclusion from the critical region on 8p. Am J Med Genet 57:483–488 - PubMed
-
- Bamford RN, Roessler E, Burdine RD, Saplakoglu U, dela Cruz J, Splitt M, Goodship JA, Towbin J, Bowers P, Ferrero GB, Marino B, Schier AF, Shen MM, Muenke M, Casey B (2000) Loss-of-function mutations in the EGF-CFC gene CFC1 are associated with human left-right laterality defects. Nat Genet 26:365–369 - PubMed
-
- Barlow GM, Chen XN, Shi ZY, Lyons GE, Kurnit DM, Celle L, Spinner NB, Zackai E, Pettenati MJ, Van Riper AJ, Vekemans MJ, Mjaatvedt CH, Korenberg JR (2001) Down syndrome congenital heart disease: a narrowed region and a candidate gene. Genet Med 3:91–101 - PubMed
-
- Collod-Beroud G, Beroud C, Ades L, Black C, Boxer M, Brocks DJH, Holman KJ, de Paepe A, Francke U, Grau U, Hayward C, Klein HG, Liu WG, Nuytinck L, Peltonen L, Perez ABA, Rantamaki T, Junien C, Boileau C (1998) Marfan database (3rd ed): new mutations and new routines for the software. Nucleic Acids Research 26:229–233 - PMC - PubMed
-
- Cousineau AJ, Lauer RM, Pierpont ME, Burns TL, Ardinger RH, Patil SR, Sheffield VC (1994) Linkage analysis of autosomal dominant atrioventricular canal defects: exclusion of chromosome 21. Hum Genet 93:103–108 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
