

Tumor necrosis factor alpha inhibition of hepatitis B virus replication involves disruption of capsid Integrity through activation of NF-kappaB
- PMID: 12634363
- PMCID: PMC150632
- DOI: 10.1128/jvi.77.7.4033-4042.2003
Tumor necrosis factor alpha inhibition of hepatitis B virus replication involves disruption of capsid Integrity through activation of NF-kappaB
Abstract
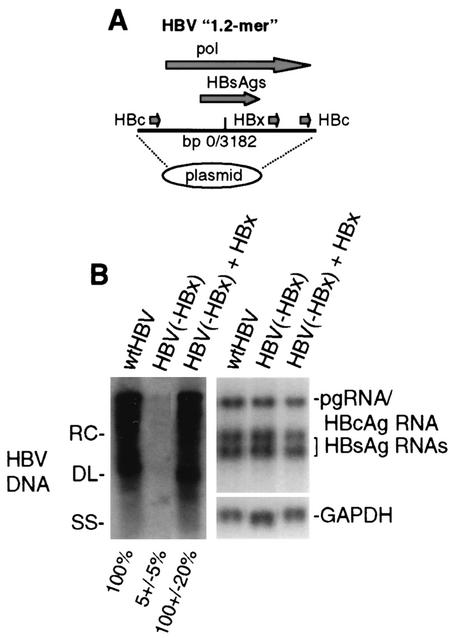
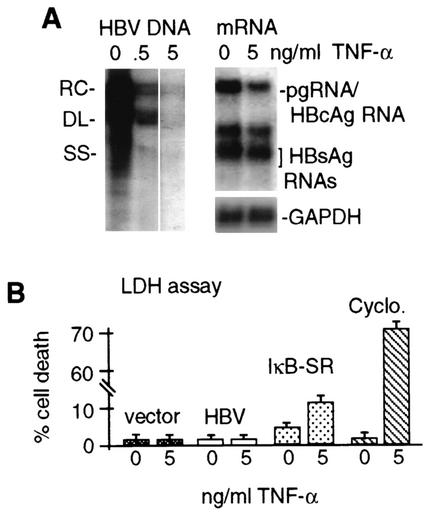
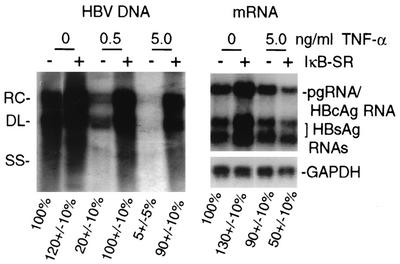
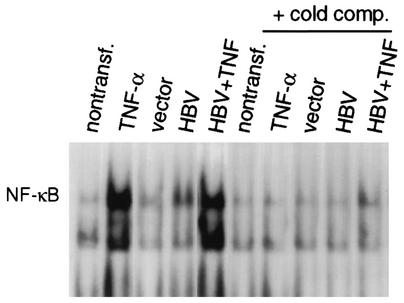
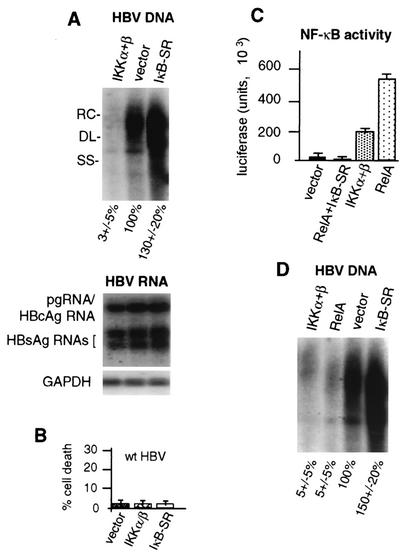
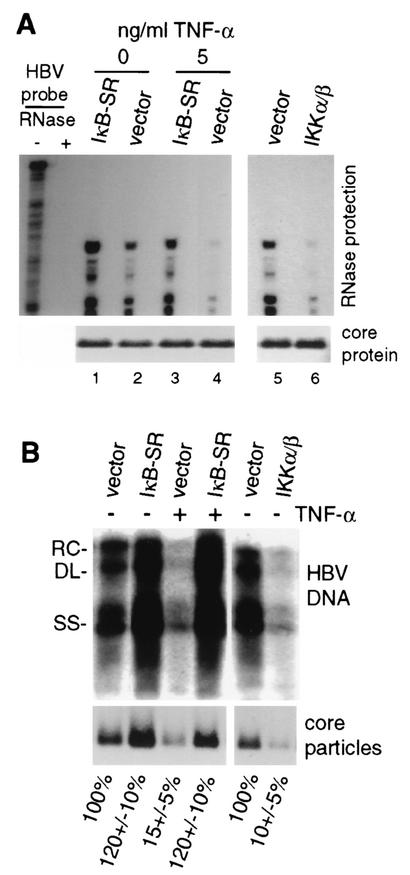
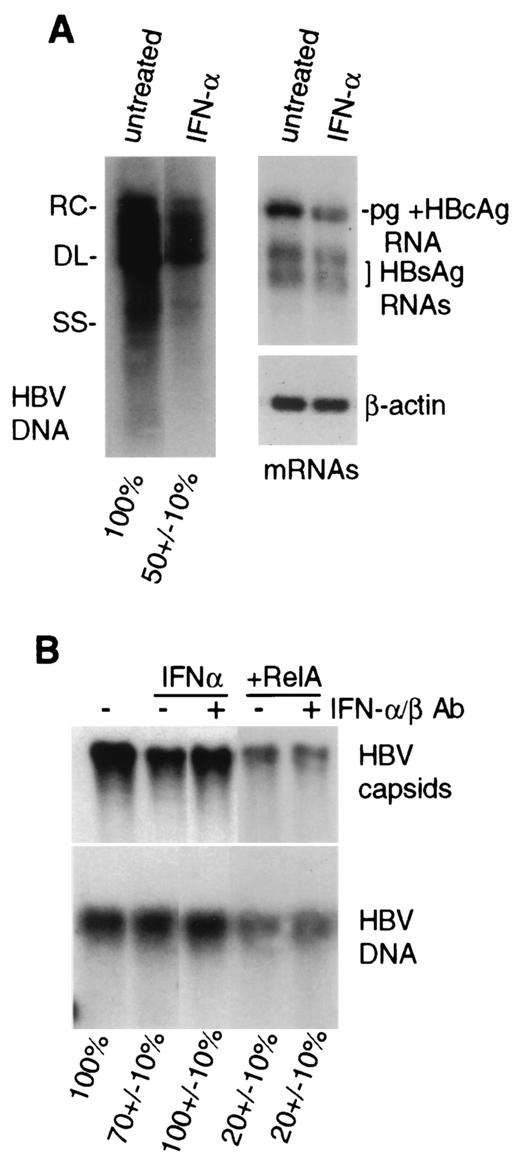
Chronic infection by hepatitis B virus results from an inability to clear the virus, which is associated with liver disease and liver cancer. Tumor necrosis factor alpha (TNF-alpha) is associated with noncytopathic clearance of hepatitis B virus in animal models. Here we demonstrate that the nuclear factor kappaB (NF-kappaB) signaling pathway is a central mediator of inhibition of hepatitis B virus by TNF-alpha and we describe the molecular mechanism. TNF-alpha is shown to suppress hepatitis B virus DNA replication without cell killing by disrupting the formation or stability of cytoplasmic viral capsids through a pathway requiring the NF-kappaB-activating inhibitor of kappaB kinase complex IKK-alpha/beta and active transcription factor NF-kappaB. Hepatitis B virus replication could also be inhibited and viral capsid formation could be disrupted in the absence of TNF-alpha solely by overexpression of IKK-alpha/beta or strong activation of NF-kappaB. In contrast, inhibition of NF-kappaB signaling stimulated viral replication, demonstrating that HBV replication is both positively and negatively regulated by the level of activity of the NF-kappaB pathway. Studies are presented that exclude the possibility that HBV inhibition by NF-kappaB is carried out by secondary production of gamma interferon or alpha/beta interferon. These results identify a novel mechanism for noncytopathic suppression of hepatitis B virus replication that is mediated by the NF-kappaB signaling pathway and activated by TNF-alpha.
Figures
References
-
- Beg, A. A., W. C. Sha, R. T. Bronson, S. Ghosh, and D. Baltimore. 1995. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-κB. Nature 376:167-170. - PubMed
-
- Bergametti, F., S. Prigent, B. Luber, A. Benoit, P. Tiollais, A. Sarasin, and C. Transy. 1999. The proapoptotic effect of hepatitis B virus HBx protein correlates with its transactivation activity in stably transfected cell lines. Oncogene 18:2860-2871. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
