

Histone deacetylase inhibitors prevent oxidative neuronal death independent of expanded polyglutamine repeats via an Sp1-dependent pathway
- PMID: 12640146
- PMCID: PMC153084
- DOI: 10.1073/pnas.0737363100
Histone deacetylase inhibitors prevent oxidative neuronal death independent of expanded polyglutamine repeats via an Sp1-dependent pathway
Erratum in
- Proc Natl Acad Sci U S A. 2003 May 27;100(11):6890. Deodoglu, A [corrected to Dedeoglu, A]
Abstract
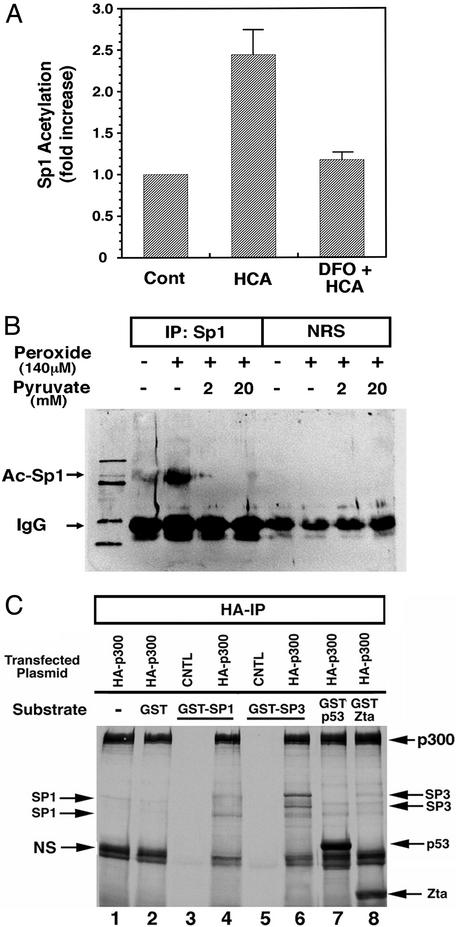
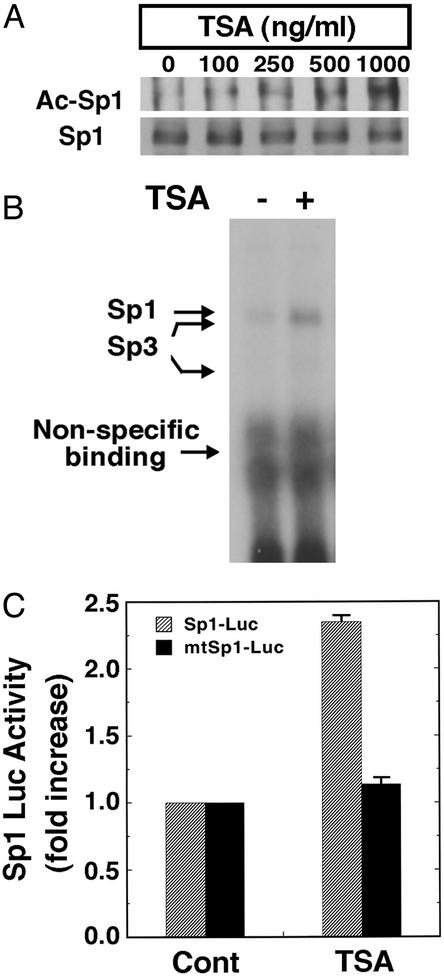
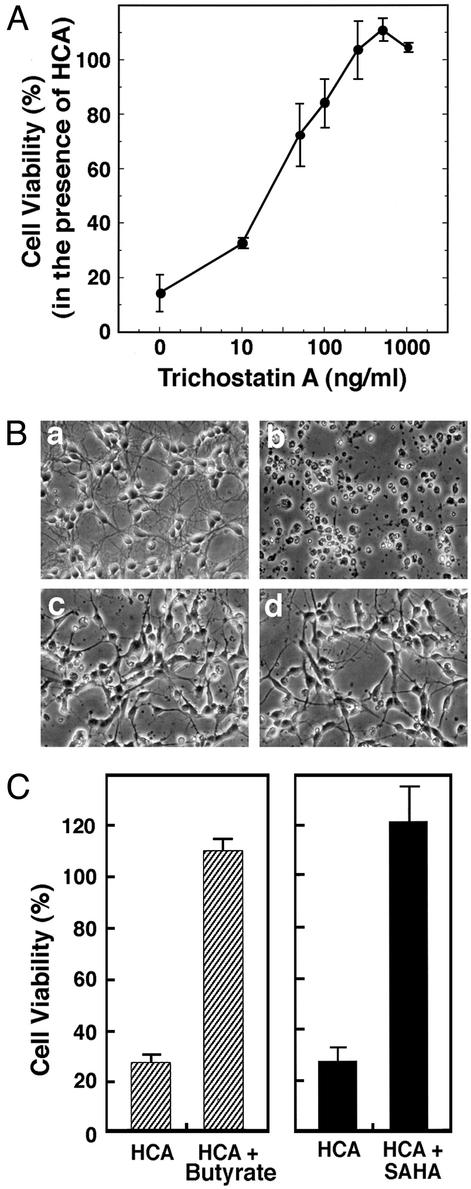
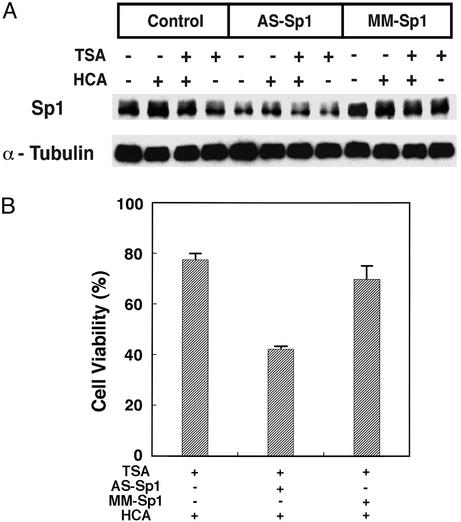
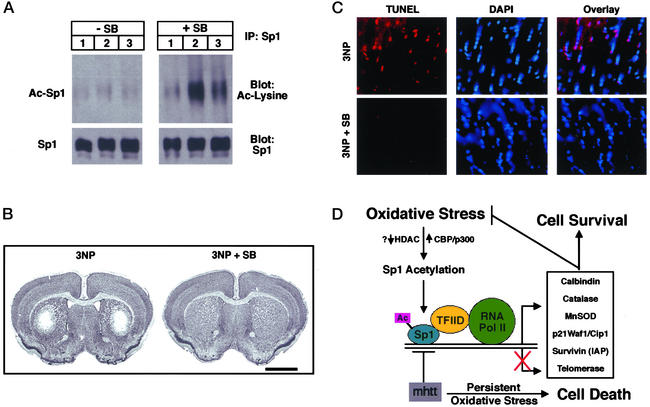
Oxidative stress is believed to be an important mediator of neurodegeneration. However, the transcriptional pathways induced in neurons by oxidative stress that activate protective gene responses have yet to be fully delineated. We report that the transcription factor Sp1 is acetylated in response to oxidative stress in neurons. Histone deacetylase (HDAC) inhibitors augment Sp1 acetylation, Sp1 DNA binding, and Sp1-dependent gene expression and confer resistance to oxidative stress-induced death in vitro and in vivo. Sp1 activation is necessary for the protective effects of HDAC inhibitors. Together, these results demonstrate that HDAC inhibitors inhibit oxidative death independent of polyglutamine expansions by activating an Sp1-dependent adaptive response.
Figures
References
-
- Sagara Y, Dargusch R, Chambers D, Davis J, Schubert D, Maher P. Free Radical Biol Med. 1998;24:1375–1389. - PubMed
-
- Atwood C S, Huang X, Moir R D, Tanzi R E, Bush A I. Met Ions Biol Syst. 1999;36:309–364. - PubMed
-
- Castagne V, Gautschi M, Lefevre K, Posada A, Clarke P G. Prog Neurobiol. 1999;59:397–423. - PubMed
-
- Estevez A G, Crow J P, Sampson J B, Reiter C, Zhuang Y, Richardson G J, Tarpey M M, Barveito L, Beckman J S. Science. 1999;286:2498–2500. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
