

Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants
- PMID: 12642658
- PMCID: PMC153051
- DOI: 10.1073/pnas.0737556100
Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants
Abstract
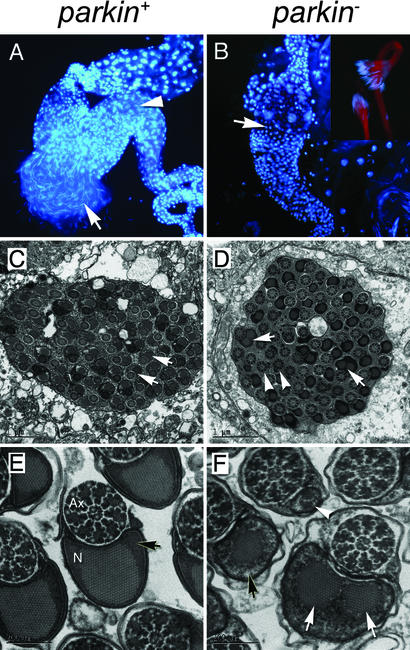
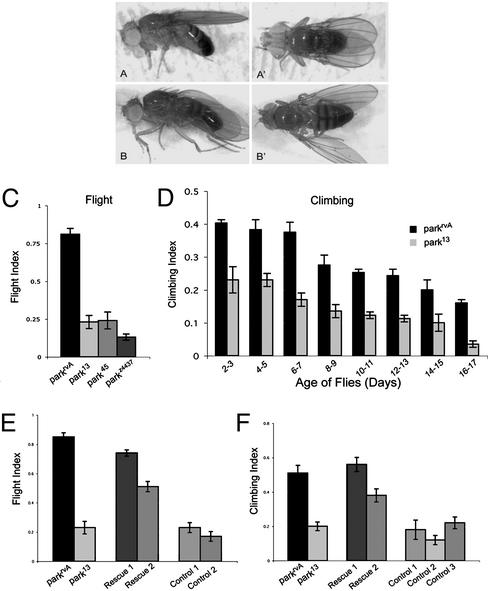
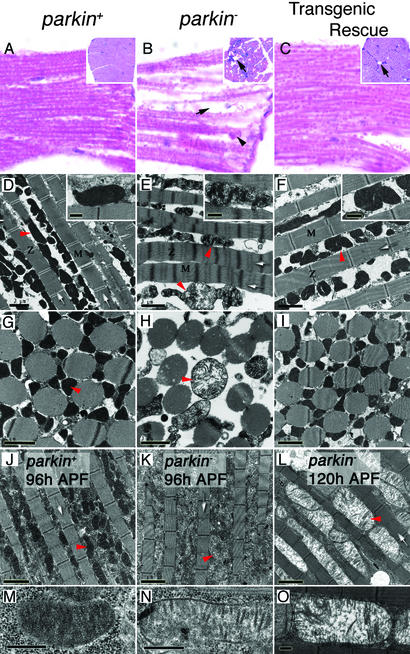
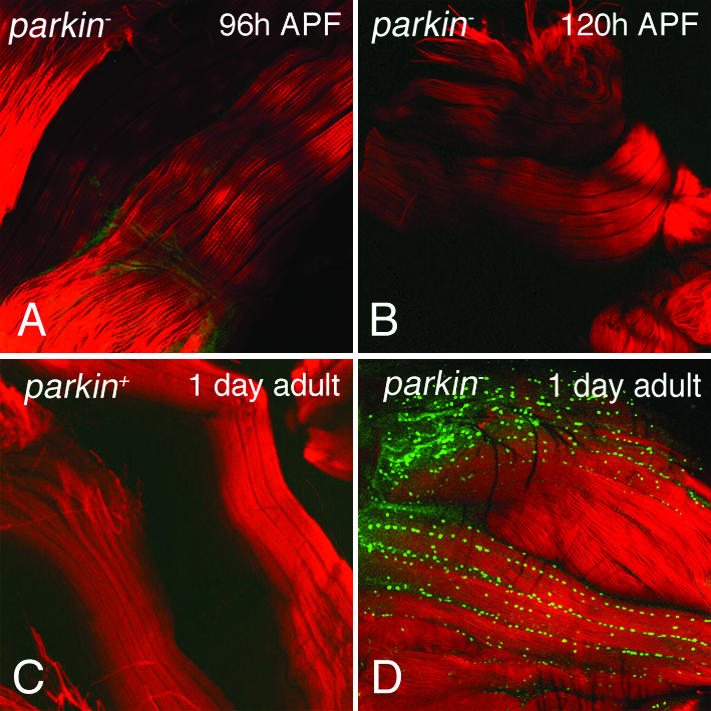
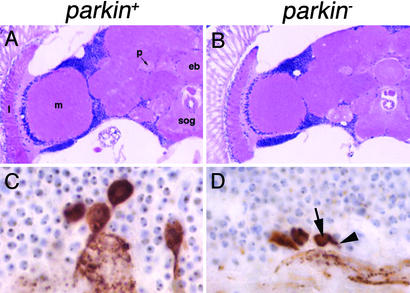
Parkinson's disease (PD) is a common neurodegenerative disorder characterized by loss of dopaminergic neurons in the substantia nigra. Several lines of evidence strongly implicate mitochondrial dysfunction as a major causative factor in PD, although the molecular mechanisms responsible for mitochondrial dysfunction are poorly understood. Recently, loss-of-function mutations in the parkin gene, which encodes a ubiquitin-protein ligase, were found to underlie a familial form of PD known as autosomal recessive juvenile parkinsonism (AR-JP). To gain insight into the molecular mechanism responsible for selective cell death in AR-JP, we have created a Drosophila model of this disorder. Drosophila parkin null mutants exhibit reduced lifespan, locomotor defects, and male sterility. The locomotor defects derive from apoptotic cell death of muscle subsets, whereas the male sterile phenotype derives from a spermatid individualization defect at a late stage of spermatogenesis. Mitochondrial pathology is the earliest manifestation of muscle degeneration and a prominent characteristic of individualizing spermatids in parkin mutants. These results indicate that the tissue-specific phenotypes observed in Drosophila parkin mutants result from mitochondrial dysfunction and raise the possibility that similar mitochondrial impairment triggers the selective cell loss observed in AR-JP.
Figures

References
-
- Vaughan J R, Davis M B, Wood N W. Ann Hum Genet. 2001;65:111–126. - PubMed
-
- Gorell J, Johnson C, Rybicki B, Peterson E, Richardson R. Neurology. 1998;50:1346–1350. - PubMed
-
- Gwinn-Hardy K A. Movement Disorders. 2002;14:645–656. - PubMed
-
- Mizuno Y, Saitoh T, Sone N. Biochem Biophys Res Commun. 1987;143:294–299. - PubMed
-
- Nicklas W, Vyas I, Heikkila R. Life Sci. 1985;36:2503–2508. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
