

Missense mutations in the regulatory domain of PKC gamma: a new mechanism for dominant nonepisodic cerebellar ataxia
- PMID: 12644968
- PMCID: PMC1180348
- DOI: 10.1086/373883
Missense mutations in the regulatory domain of PKC gamma: a new mechanism for dominant nonepisodic cerebellar ataxia
Abstract
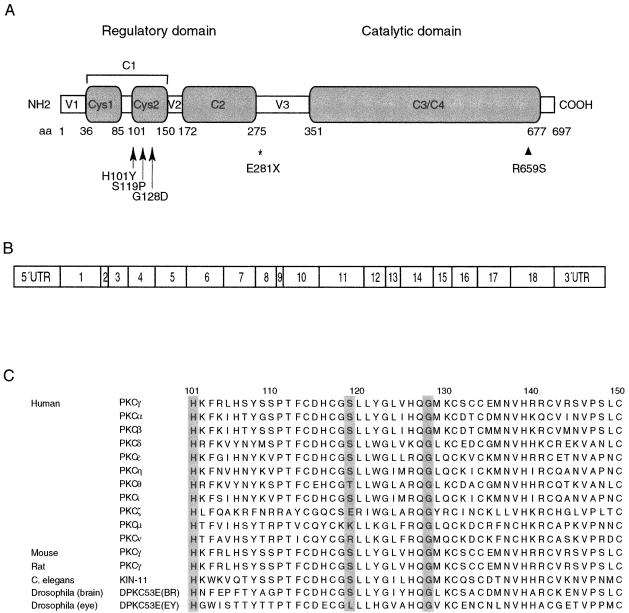
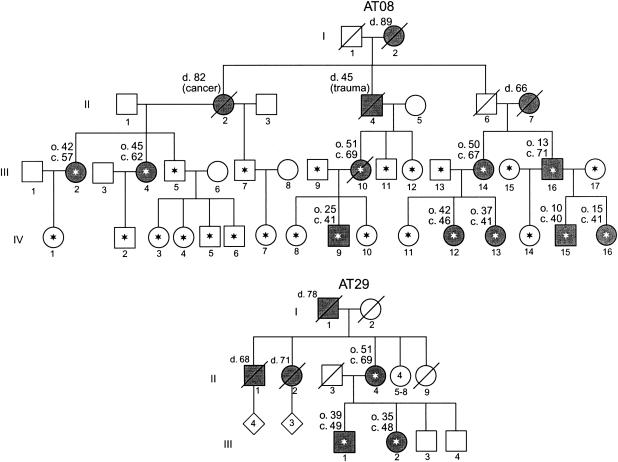
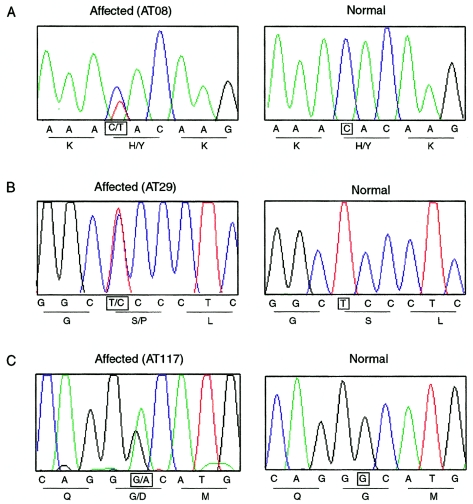
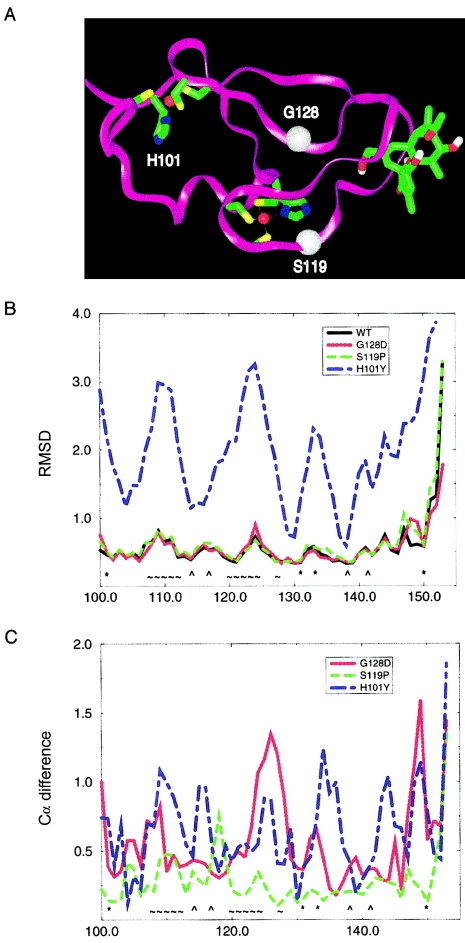
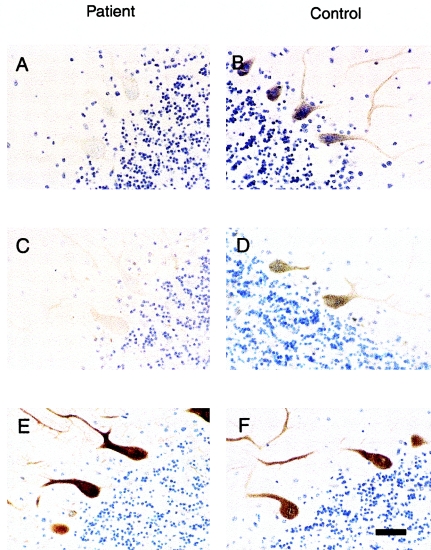
We report a nonepisodic autosomal dominant (AD) spinocerebellar ataxia (SCA) not caused by a nucleotide repeat expansion that is, to our knowledge, the first such SCA. The AD SCAs currently comprise a group of > or =16 genetically distinct neurodegenerative conditions, all characterized by progressive incoordination of gait and limbs and by speech and eye-movement disturbances. Six of the nine SCAs for which the genes are known result from CAG expansions that encode polyglutamine tracts. Noncoding CAG, CTG, and ATTCT expansions are responsible for three other SCAs. Approximately 30% of families with SCA do not have linkage to the known loci. We recently mapped the locus for an AD SCA in a family (AT08) to chromosome 19q13.4-qter. A particularly compelling candidate gene, PRKCG, encodes protein kinase C gamma (PKC gamma), a member of a family of serine/threonine kinases. The entire coding region of PRKCG was sequenced in an affected member of family AT08 and in a group of 39 unrelated patients with ataxia not attributable to trinucleotide expansions. Three different nonconservative missense mutations in highly conserved residues in C1, the cysteine-rich region of the protein, were found in family AT08, another familial case, and a sporadic case. The mutations cosegregated with disease in both families. Structural modeling predicts that two of these amino acid substitutions would severely abrogate the zinc-binding or phorbol ester-binding capabilities of the protein. Immunohistochemical studies on cerebellar tissue from an affected member of family AT08 demonstrated reduced staining for both PKC gamma and ataxin 1 in Purkinje cells, whereas staining for calbindin was preserved. These results strongly support a new mechanism for neuronal cell dysfunction and death in hereditary ataxias and suggest that there may be a common pathway for PKC gamma-related and polyglutamine-related neurodegeneration.
Figures
References
Electronic-Database Information
-
- Entrez Genome View, http://www.ncbi.nlm.nih.gov/mapview/map_search.cgi?chr=hum_chr.inf (for NCBI Map Viewer)
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for PRKCG mRNA sequence [accession number NM_002739])
-
- GeneTests Home Page, http://www.geneclinics.org/ or http://www.genetests.org/ (for “Hereditary Ataxia Overview,” by T.D.B., in GeneReviews)
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for PRKCG) - PubMed
-
- SMART, http://smart.embl-heidelberg.de/ (for domain prediction within PKCγ)
References
-
- Abeliovich A, Paylor R, Chen C, Kim JJ, Wehner JM, Tonegawa S (1993) PKC γ mutant mice exhibit mild deficits in spatial and contextual learning. Cell 75:1263–1271 - PubMed
-
- Barmack NH, Qian Z, Yoshimura J (2000) Regional and cellular distribution of protein kinase C in rat cerebellar Purkinje cells. J Comp Neurol 427:235–254 - PubMed
-
- Brkanac Z, Bylenok L, Fernandez M, Matsushita M, Lipe H, Wolff, J, Nochlin D, Raskind WH, Bird TD (2002) A new dominant spinocerebellar ataxia is linked to chromosome 19q13.4. Arch Neurol 59:1291–1295 - PubMed
-
- Burright EN, Clark HB, Servadio A, Matilla T, Feddersen RM, Yunis WS, Duvick LA, Zoghbi HY, Orr HT (1995) SCA1 transgenic mice: a model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cell 82:937–948 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
