

Mutant huntingtin causes context-dependent neurodegeneration in mice with Huntington's disease
- PMID: 12657678
- PMCID: PMC6742008
- DOI: 10.1523/JNEUROSCI.23-06-02193.2003
Mutant huntingtin causes context-dependent neurodegeneration in mice with Huntington's disease
Abstract
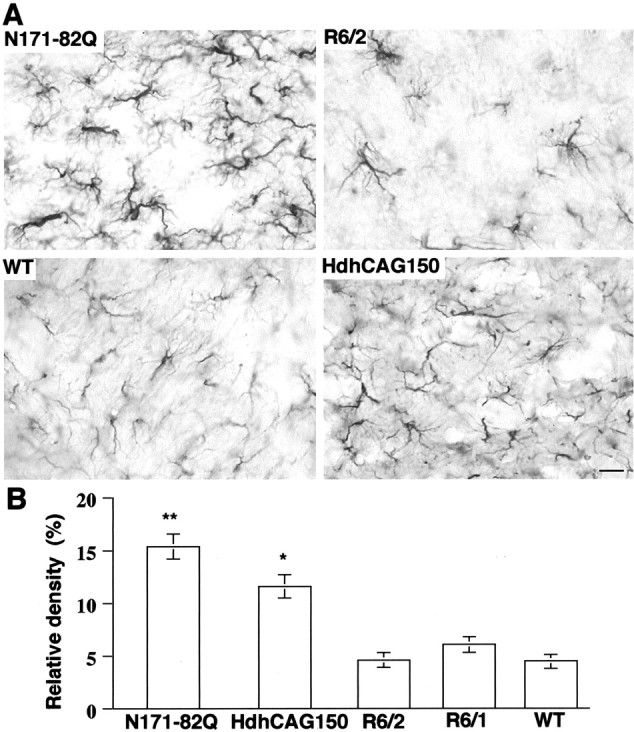
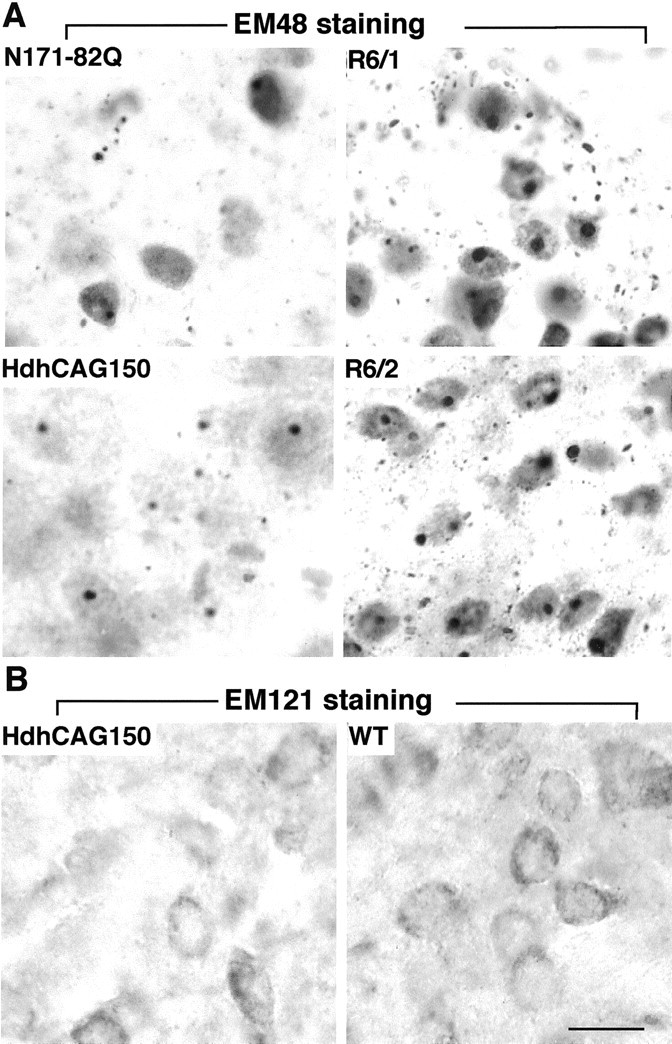
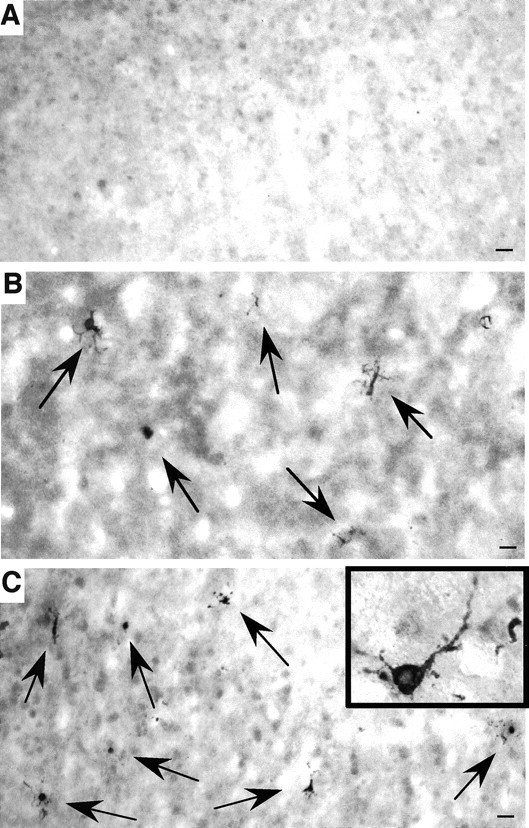
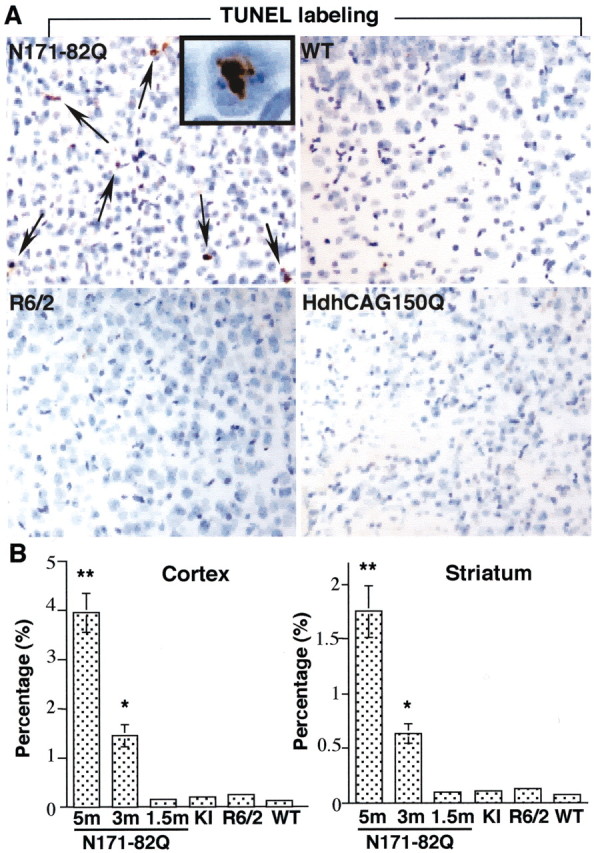
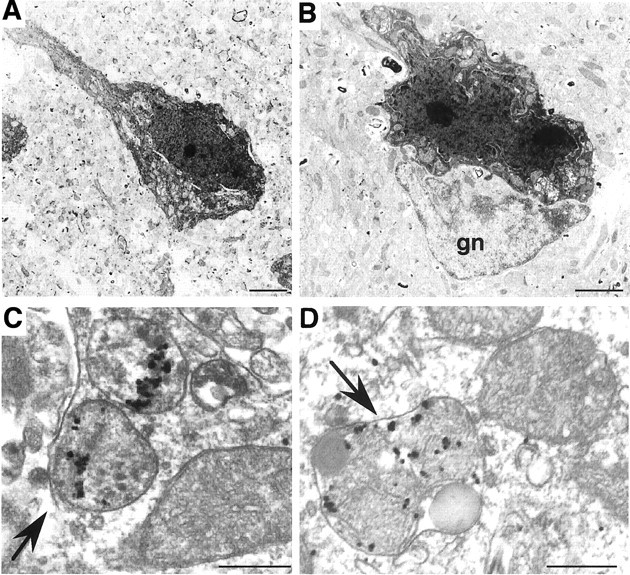
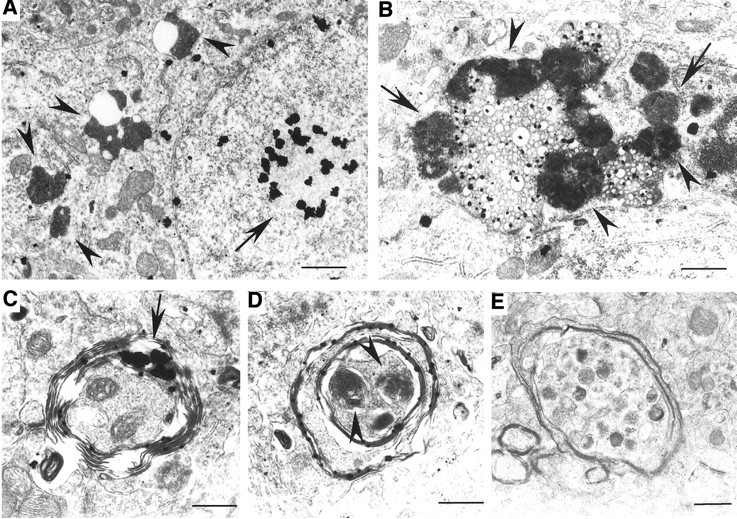
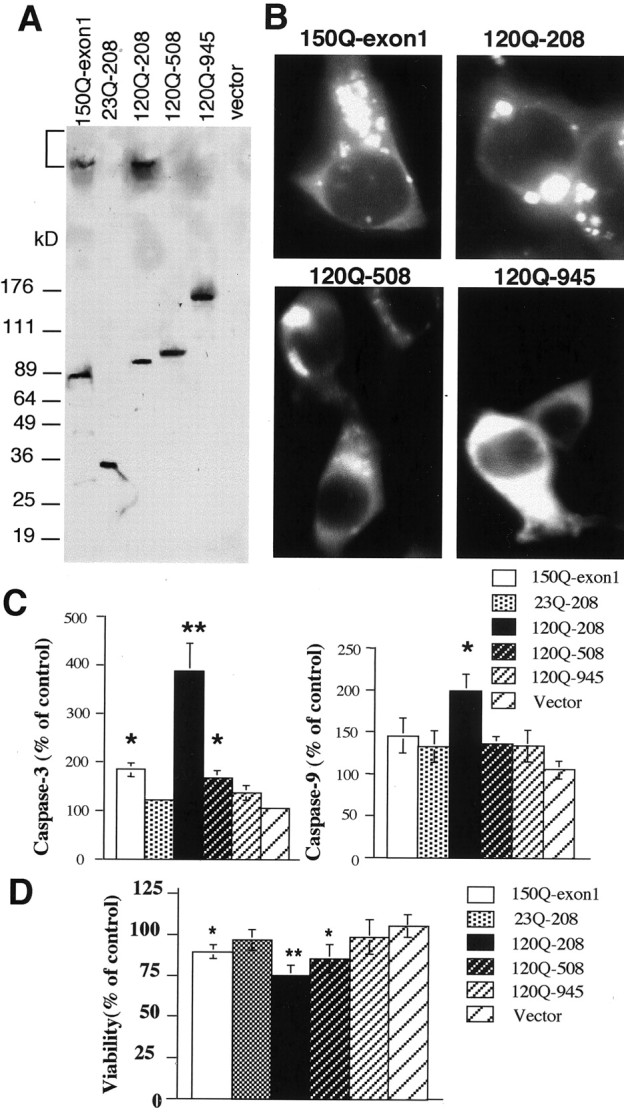
Huntington's disease (HD) mouse models that express N-terminal huntingtin fragments show rapid disease progression and have been used for developing therapeutics. However, light microscopy reveals no significant neurodegeneration in these mice. It remains unclear how mutant huntingtin induces neurodegeneration. Using caspase staining, terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling, and electron microscopy, we observed that N171-82Q mice, which express the first 171 aa of mutant huntingtin, displayed more degenerated neurons than did other HD mouse models. The neurodegeneration was also evidenced by increased immunostaining for glial fibrillary acidic protein and ultrastructural features of apoptosis. R6/2 mice, which express exon 1 of mutant huntingtin, showed dark, nonapoptotic neurons and degenerated mitochondria associated with mutant huntingtin. In HD repeat knock-in mice (HdhCAG150), which express full-length mutant huntingtin, degenerated cytoplasmic organelles were found in both axons and neuronal cell bodies in association with mutant huntingtin that was not labeled by an antibody to huntingtin amino acids 342-456. Transfection of cultured cells with mutant huntingtin revealed that an N-terminal huntingtin fragment (amino acids 1-208 plus a 120 glutamine repeat) caused a greater increase in caspase activity than did exon 1 huntingtin and longer huntingtin fragments. These results suggest that context-dependent neurodegeneration in HD may be mediated by different N-terminal huntingtin fragments. In addition, this study has identified neurodegenerative markers for the evaluation of therapeutic treatments in HD mouse models.
Figures

References
-
- Andreassen OA, Dedeoglu A, Ferrante RJ, Jenkins BG, Ferrante KL, Thomas M, Friedlich A, Browne SE, Schilling G, Borchelt DR, Hersch SM, Ross CA, Beal MF. Creatine increases survival and delays motor symptoms in a transgenic animal model of Huntington's disease. Neurobiol Dis. 2001;8:479–491. - PubMed
-
- Beal MF. Energetics in the pathogenesis of neurodegenerative diseases. Trends Neurosci. 2000;23:298–304. - PubMed
-
- Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292:1552–1555. - PubMed
-
- Chan EY, Luthi-Carter R, Strand A, Solano SM, Hanson SA, DeJohn MM, Kooperberg C, Chase KO, DiFiglia M, Young AB, Leavitt BR, Cha JH, Aronin N, Hayden MR, Olson JM. Increased huntingtin protein length reduces the number of polyglutamine-induced gene expression changes in mouse models of Huntington's disease. Hum Mol Genet. 2002;11:1939–1951. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases