

Molecular analysis of hereditary nonpolyposis colorectal cancer in the United States: high mutation detection rate among clinically selected families and characterization of an American founder genomic deletion of the MSH2 gene
- PMID: 12658575
- PMCID: PMC1180263
- DOI: 10.1086/373963
Molecular analysis of hereditary nonpolyposis colorectal cancer in the United States: high mutation detection rate among clinically selected families and characterization of an American founder genomic deletion of the MSH2 gene
Abstract
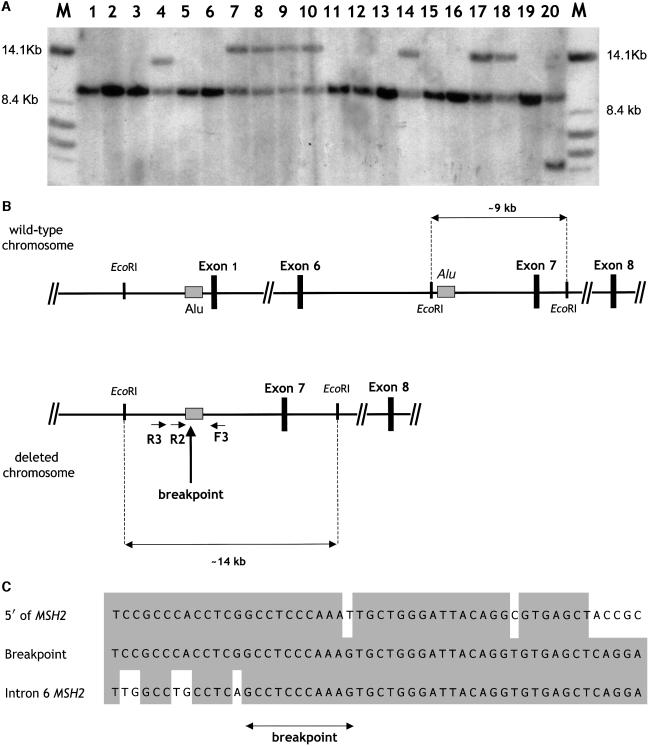
The identification of germline mutations in families with HNPCC is hampered by genetic heterogeneity and clinical variability. In previous studies, MSH2 and MLH1 mutations were found in approximately two-thirds of the Amsterdam-criteria-positive families and in much lower percentages of the Amsterdam-criteria-negative families. Therefore, a considerable proportion of HNPCC seems not to be accounted for by the major mismatch repair (MMR) genes. Does the latter result from a lack of sensitivity of mutation detection techniques, or do additional genes underlie the remaining cases? In this study we address these questions by thoroughly investigating a cohort of clinically selected North American families with HNPCC. We analyzed 59 clinically well-defined U.S. families with HNPCC for MSH2, MLH1, and MSH6 mutations. To maximize mutation detection, different techniques were employed, including denaturing gradient gel electrophoresis, Southern analysis, microsatellite instability, immunohistochemistry, and monoallelic expression analysis. In 45 (92%) of the 49 Amsterdam-criteria-positive families and in 7 (70%) of the 10 Amsterdam-criteria-negative families, a mutation was detected in one of the three analyzed MMR genes. Forty-nine mutations were in MSH2 or MLH1, and only three were in MSH6. A considerable proportion (27%) of the mutations were genomic rearrangements (12 in MSH2 and 2 in MLH1). Notably, a deletion encompassing exons 1-6 of MSH2 was detected in seven apparently unrelated families (12% of the total cohort) and was subsequently proven to be a founder. Screening of a second U.S. cohort with HNPCC from Ohio allowed the identification of two additional kindreds with the identical founder deletion. In the present study, we show that optimal mutation detection in HNPCC is achieved by combining accurate and expert clinical selection with an extensive mutation detection strategy. Notably, we identified a common North American deletion in MSH2, accounting for approximately 10% of our cohort. Genealogical, molecular, and haplotype studies showed that this deletion represents a North American founder mutation that could be traced back to the 19th century.
Figures

References
Electronic-Database Information
-
- FamilySearch Internet, http://www.familysearch.org/Eng/Search/frameset_search.asp
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for HNPCC) - PubMed
-
- RepeatMasker Web Server, http://ftp.genome.Washington.edu/cgi-bin/RepeatMasker
References
-
- Aaltonen LA, Peltomaki P, Mecklin JP, Jarvinen H, Jass JR, Green S, Lynch HT, Watson P, Tallqvist G, Juhola M (1994) Replication errors in benign and malignant tumors from hereditary nonpolyposis colorectal cancer patients. Cancer Res 54:1645–1648 - PubMed
-
- Aarnio M, Sankila R, Pukkala E, Salovaara R, Aaltonen LA, de la Chapelle A, Peltomaki P, Mecklin JP, Jarvinen HJ (1999) Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer 81:214–218 - PubMed
-
- Akiyama Y, Sato H, Yamada T, Nagasaki H, Tsuchiya A, Abe R, Yuasa Y (1997) Germ-line mutation of the hMSH6/GTBP gene in an atypical hereditary nonpolyposis colorectal cancer kindred. Cancer Res 57:3920–3923 - PubMed
-
- Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, Srivastava S (1998) A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 58:5248–5257 - PubMed
-
- Bronner CE, Baker SM, Morrison PT, Warren G, Smith LG, Lescoe MK, Kane M, Earabino C, Lipford J, Lindblom A, Tannergård P, Bollag R, Godwin A, Ward DC, Nordenskjöld M, Fishel R, Kolodner R, Liskay M (1994) Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature 368:258–261 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
