

Human Tousled like kinases are targeted by an ATM- and Chk1-dependent DNA damage checkpoint
- PMID: 12660173
- PMCID: PMC152895
- DOI: 10.1093/emboj/cdg151
Human Tousled like kinases are targeted by an ATM- and Chk1-dependent DNA damage checkpoint
Abstract
All eukaryotes respond to DNA damage by modulation of diverse cellular processes to preserve genomic integrity and ensure survival. Here we identify mammalian Tousled like kinases (Tlks) as a novel target of the DNA damage checkpoint. During S-phase progression, when Tlks are maximally active, generation of DNA double-strand breaks (DSBs) leads to rapid and transient inhibition of Tlk activity. Experiments with chemical inhibitors, genetic models and gene targeting through RNA interference demonstrate that this response to DSBs requires ATM and Chk1 function. Chk1 phosphorylates Tlk1 on serine 695 (S695) in vitro, and this UCN-01- and caffeine-sensitive site is phosphorylated in vivo in response to DNA damage. Substitution of S695 to alanine impaired efficient downregulation of Tlk1 after DNA damage. These findings identify an unprecedented functional co- operation between ATM and Chk1 in propagation of a checkpoint response during S phase and suggest that, through transient inhibition of Tlk kinases, the ATM-Chk1-Tlk pathway may regulate processes involved in chromatin assembly.
Figures



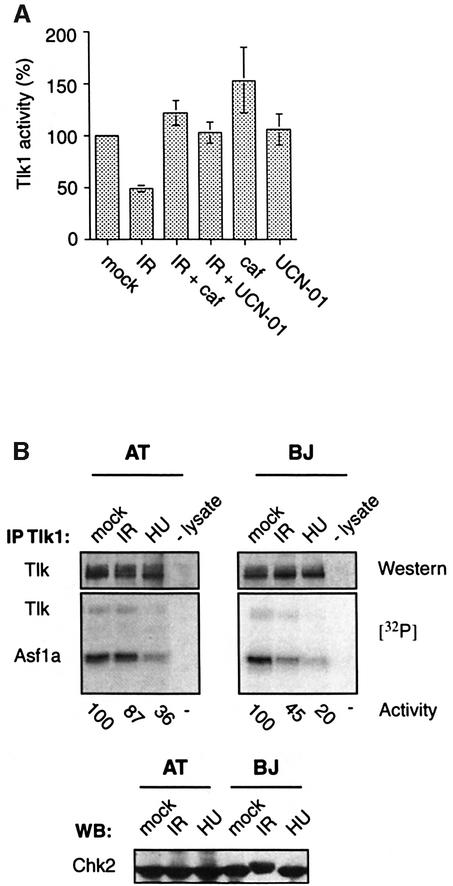
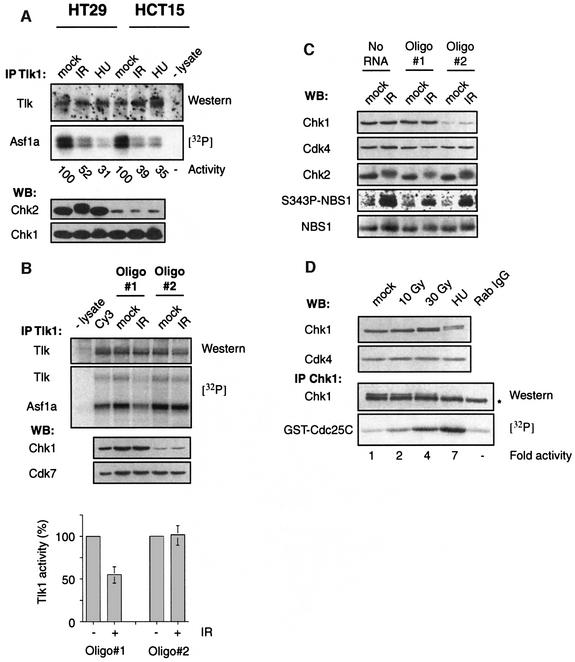

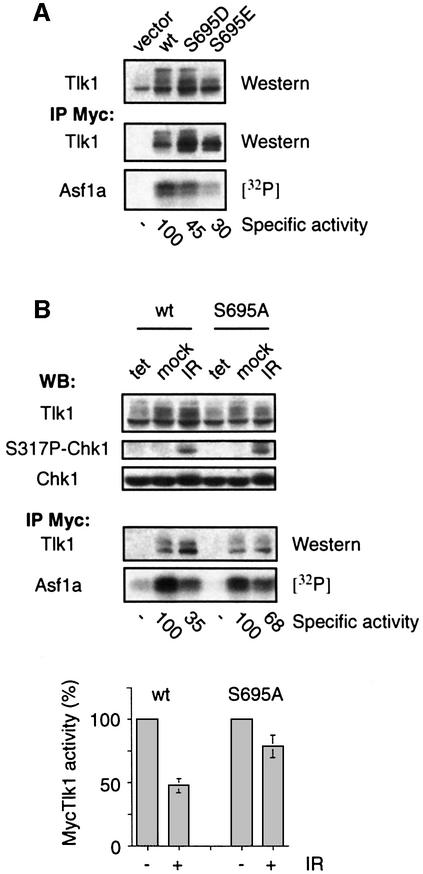
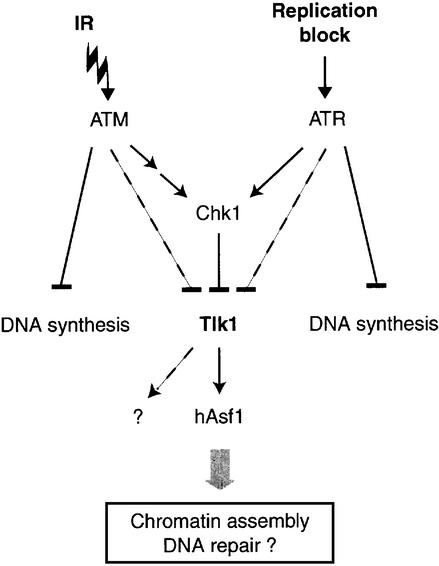
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
