

Crystal structure of human beta-hexosaminidase B: understanding the molecular basis of Sandhoff and Tay-Sachs disease
- PMID: 12662933
- PMCID: PMC2910754
- DOI: 10.1016/s0022-2836(03)00216-x
Crystal structure of human beta-hexosaminidase B: understanding the molecular basis of Sandhoff and Tay-Sachs disease
Abstract
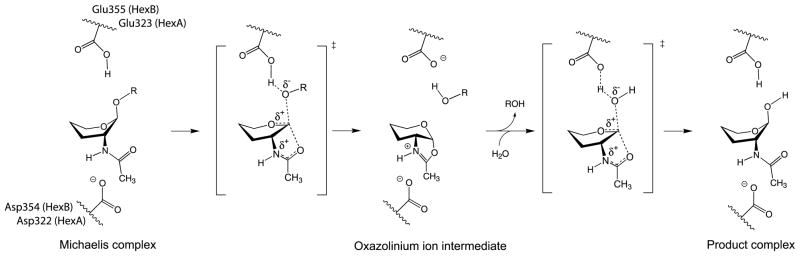
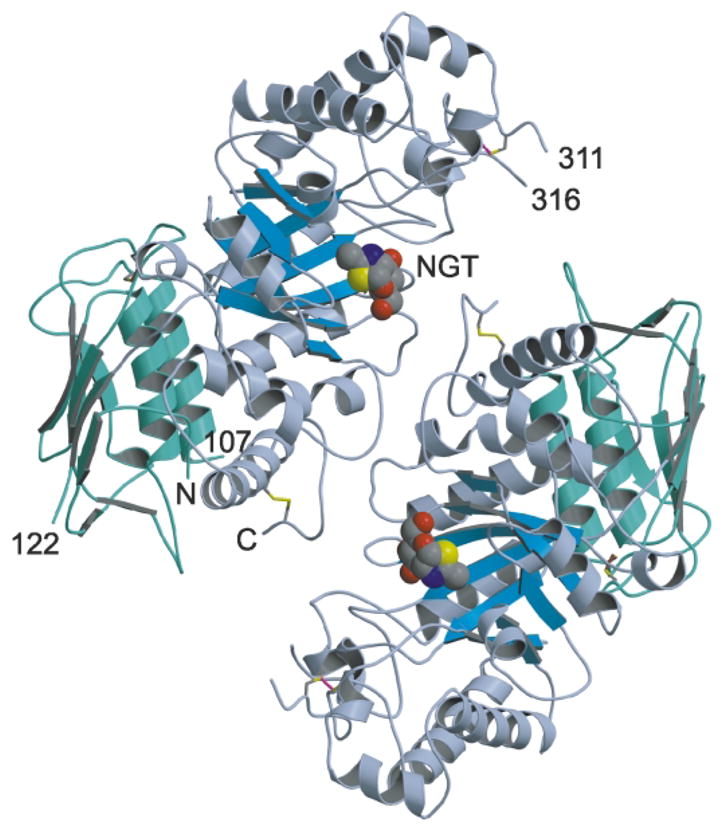
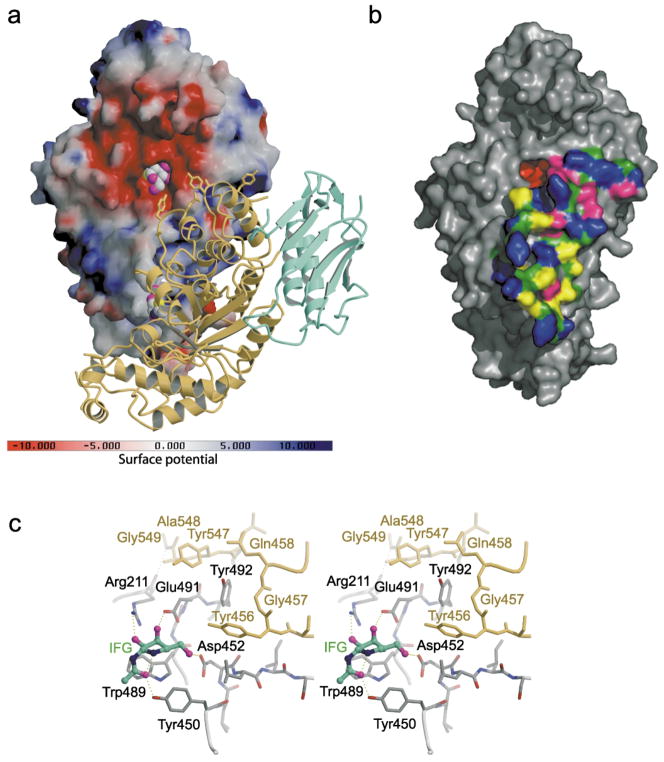
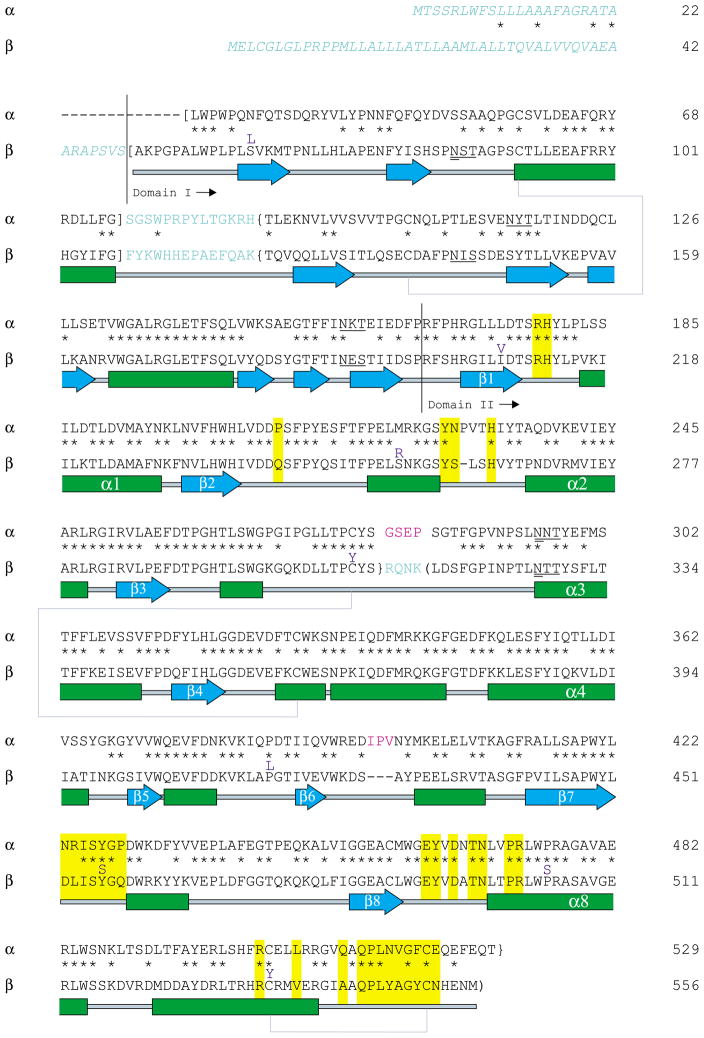
In humans, two major beta-hexosaminidase isoenzymes exist: Hex A and Hex B. Hex A is a heterodimer of subunits alpha and beta (60% identity), whereas Hex B is a homodimer of beta-subunits. Interest in human beta-hexosaminidase stems from its association with Tay-Sachs and Sandhoff disease; these are prototypical lysosomal storage disorders resulting from the abnormal accumulation of G(M2)-ganglioside (G(M2)). Hex A degrades G(M2) by removing a terminal N-acetyl-D-galactosamine (beta-GalNAc) residue, and this activity requires the G(M2)-activator, a protein which solubilizes the ganglioside for presentation to Hex A. We present here the crystal structure of human Hex B, alone (2.4A) and in complex with the mechanistic inhibitors GalNAc-isofagomine (2.2A) or NAG-thiazoline (2.5A). From these, and the known X-ray structure of the G(M2)-activator, we have modeled Hex A in complex with the activator and ganglioside. Together, our crystallographic and modeling data demonstrate how alpha and beta-subunits dimerize to form either Hex A or Hex B, how these isoenzymes hydrolyze diverse substrates, and how many documented point mutations cause Sandhoff disease (beta-subunit mutations) and Tay-Sachs disease (alpha-subunit mutations).
Figures
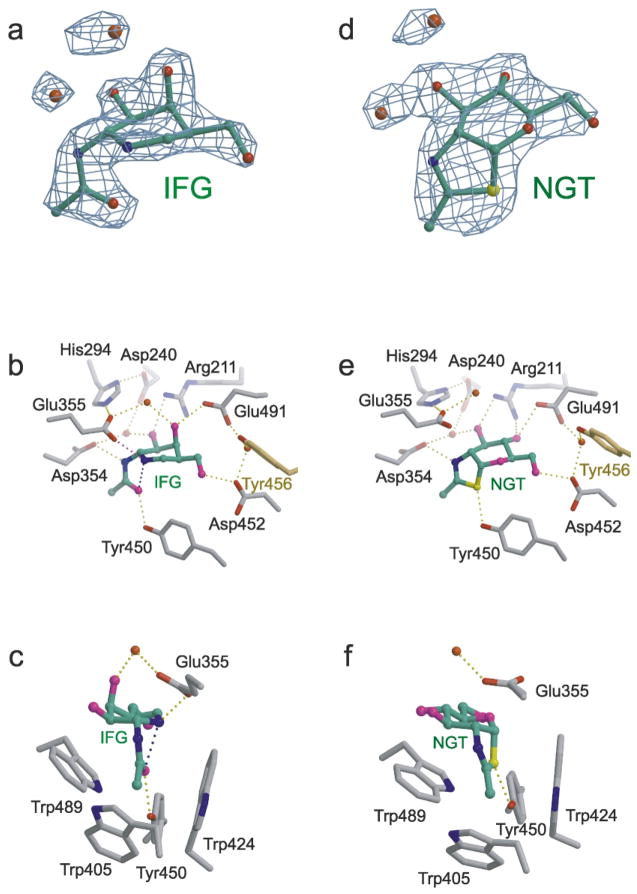
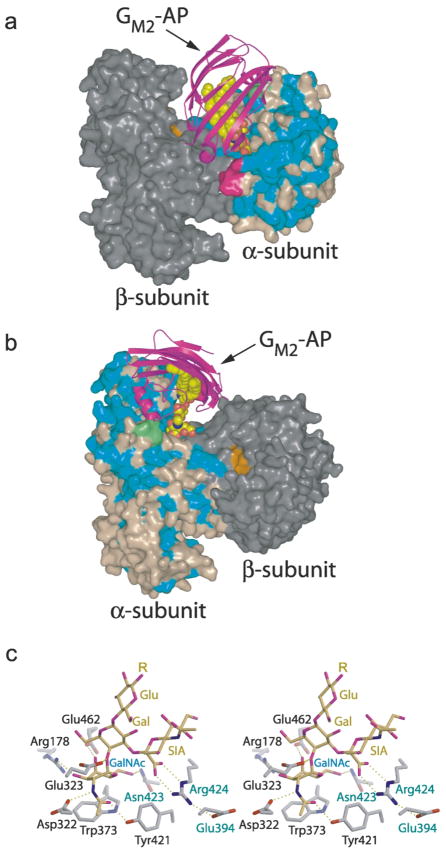
References
-
- Watanabe K. Biochemical studies on carbohydrates. J Biochem (Tokyo) 1936;24:297.
-
- Tay W. Symmetrical changes in the region of the yellow spot in each eye of an infant. Trans Ophthalmol Soc UK. 1881;1:155–157.
-
- Gravel RA, Clarke JTR, Kaback MM, Mahuran D, Sandhoff K, Suzuki K. The GM2 gangliosidoses. In: Scriver CR, Beaudet AL, Sly WS, editors. The Metabolic and Molecular Bases of Inherited Disease. 7. Vol. 2. Vol. 3 McGraw-Hill; New York: 1995. pp. 2839–2879.
-
- Mahuran DJ. Biochemical consequences of mutations causing the GM2 gangliosidoses. Biochim Biophys Acta. 1999;1455:105–138. - PubMed
-
- Mahuran DJ. The GM2 activator protein, its roles as a co-factor in GM2 hydrolysis and as a general glycolipid transport protein. Biochim Biophys Acta. 1998;1393:1–18. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
