

CXCL10 production from cytomegalovirus-stimulated microglia is regulated by both human and viral interleukin-10
- PMID: 12663757
- PMCID: PMC152158
- DOI: 10.1128/jvi.77.8.4502-4515.2003
CXCL10 production from cytomegalovirus-stimulated microglia is regulated by both human and viral interleukin-10
Abstract
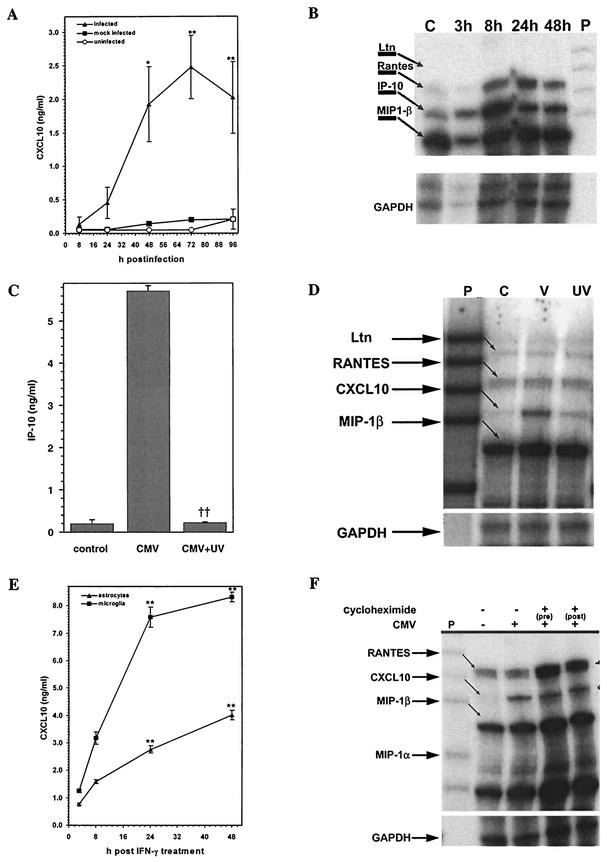
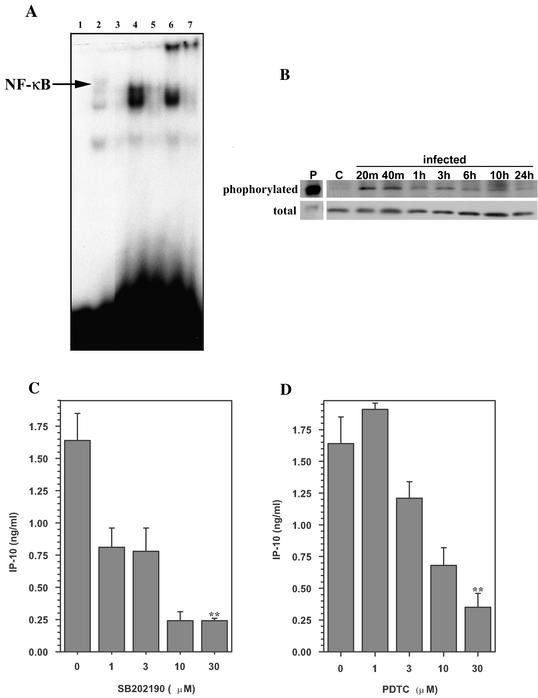
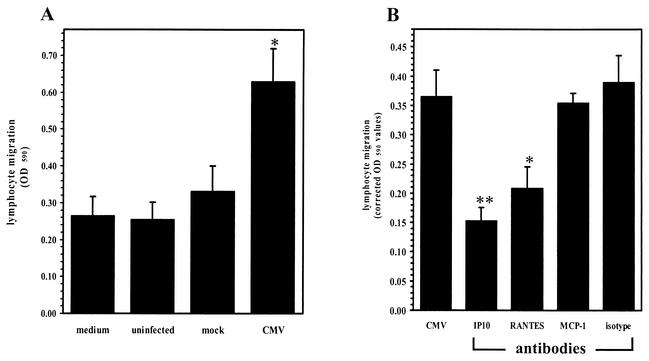
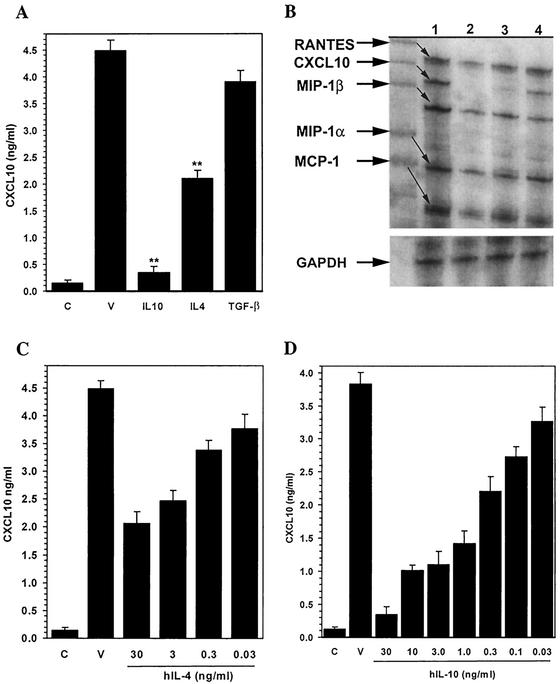
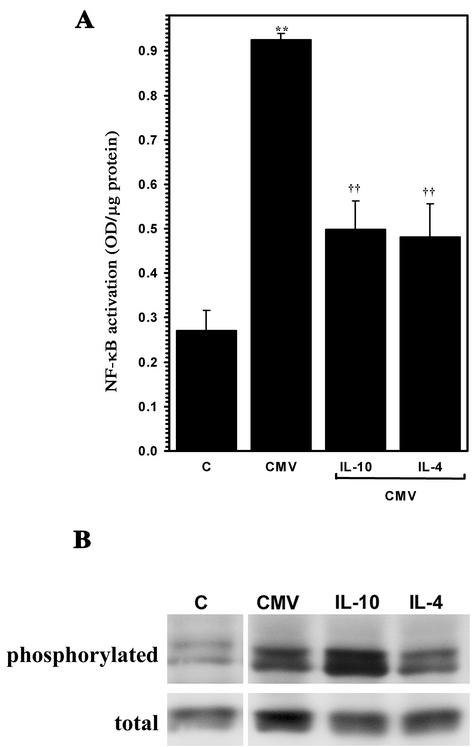
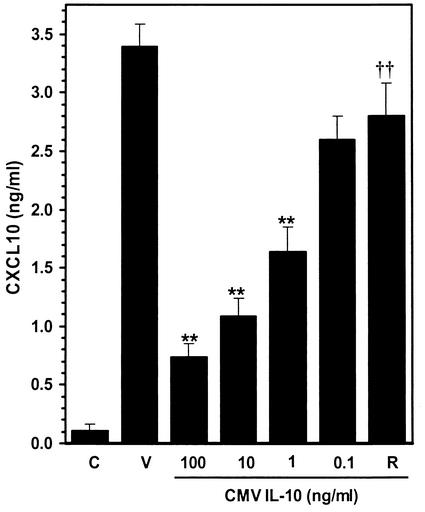
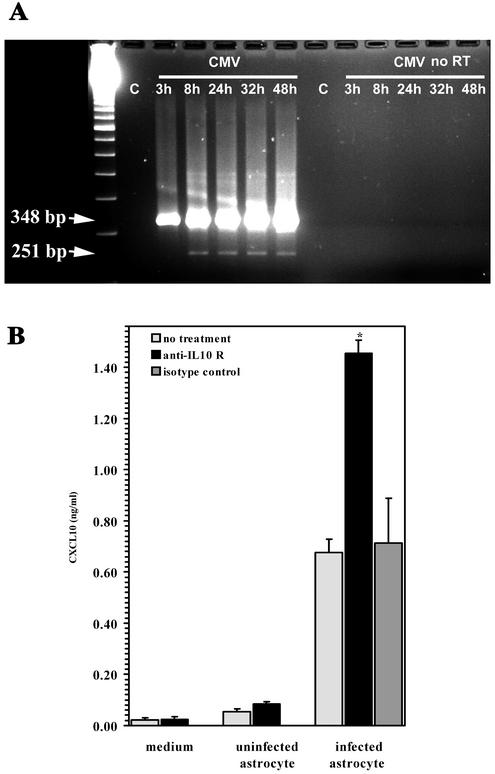
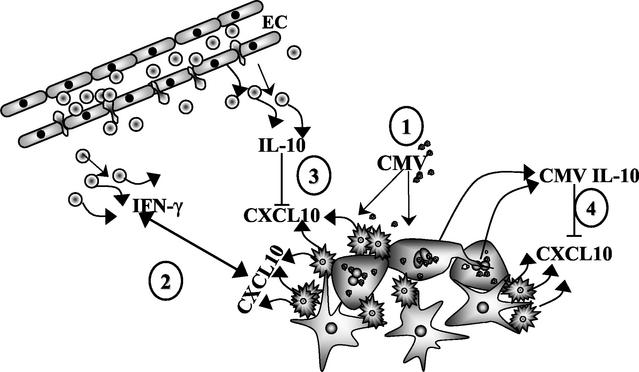
Glial cells orchestrate immunocyte recruitment to focal areas of viral infection within the brain and synchronize immune cell functions through a regulated network of cytokines and chemokines. Since recruitment of T lymphocytes plays a critical role in resolving cytomegalovirus (CMV) infection, we investigated the production of a T-cell chemoattractant, CXCL10 (gamma interferon-inducible protein 10) in response to viral infection of human glial cells. Infection with CMV was found to elicit the production of CXCL10 from primary microglial cells but not from astrocytes. This CXCL10 expression was not dependent on secondary protein synthesis but did require the phosphorylation of p38 mitogen-activated protein (MAP) kinase. In addition, migration of activated lymphocytes toward supernatants from CMV-stimulated microglial cells was partially suppressed by anti-CXCL10 antibodies. Since regulation of central nervous system inflammation is essential to allow viral clearance without immunopathology, microglial cells were then treated with anti-inflammatory cytokines. CMV-induced CXCL10 production from microglial cells was suppressed following treatment with interleukin-10 (IL-10) and IL-4 but not following treatment with transforming growth factor beta. The IL-10-mediated inhibition of CXCL10 production was associated with decreased CMV-induced NF-kappa B activation but not decreased p38 MAP kinase phosphorylation. Finally, CMV infection of fully permissive astrocytes resulted in mRNA expression for the viral homologue to human IL-10 (i.e., cmvIL-10 [UL111a]) in its spliced form and conditioned medium from CMV-infected astrocytes inhibited virus-induced CXCL10 production from microglial cells through the IL-10 receptor. These findings present yet another mechanism through which CMV may subvert host immune responses.
Figures
References
-
- Aloisi, F. 2001. Immune function of microglia. Glia 36:165-179. - PubMed
-
- Arribas, J. R., G. A. Storch, D. B. Clifford, and A. C. Tselis. 1996. Cytomegalovirus encephalitis. Ann. Intern. Med. 125:577-587. - PubMed
-
- Asensio, V. C., J. Maier, R. Milner, K. Boztug, C. Kincaid, M. Moulard, C. Phillipson, K. Lindsley, T. Krucker, H. S. Fox, and I. L. Campbell. 2001. Interferon-independent, human immunodeficiency virus type 1 gp120-mediated induction of CXCL10/IP-10 gene expression by astrocytes in vivo and in vitro. J. Virol. 75:7067-7077. - PMC - PubMed
-
- Chang, L., and M. Karin. 2001. Mammalian MAP kinase signalling cascades. Nature 410:37-40. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
