

Comparative evolutionary genomics unveils the molecular mechanism of reassignment of the CTG codon in Candida spp
- PMID: 12670996
- PMCID: PMC430169
- DOI: 10.1101/gr.811003
Comparative evolutionary genomics unveils the molecular mechanism of reassignment of the CTG codon in Candida spp
Abstract
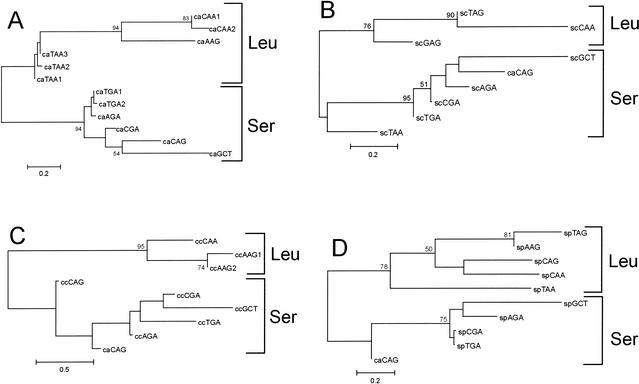
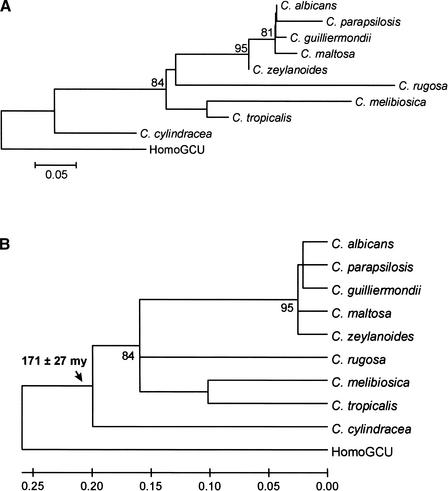
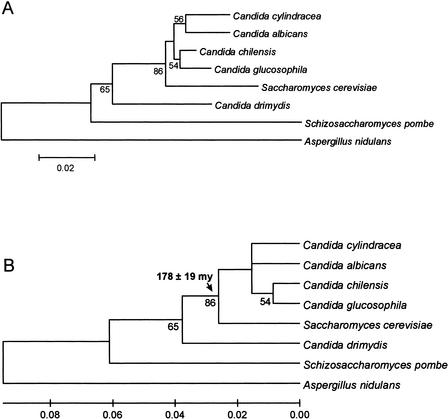
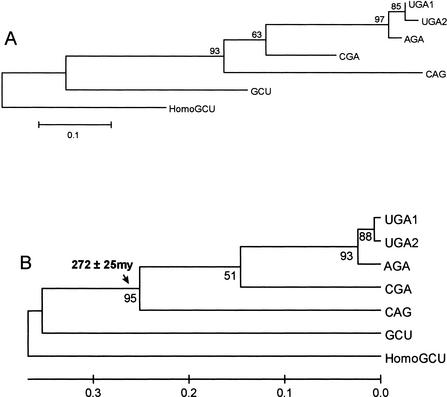
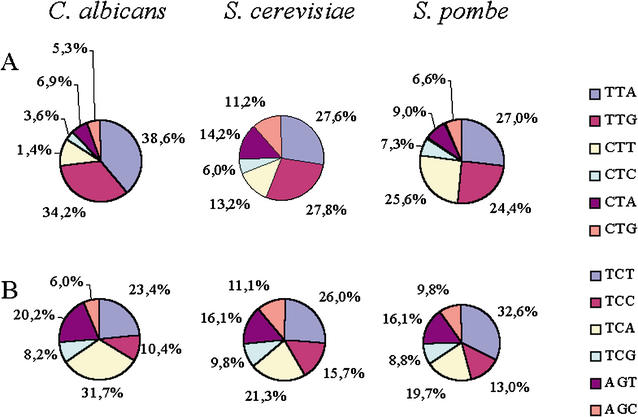
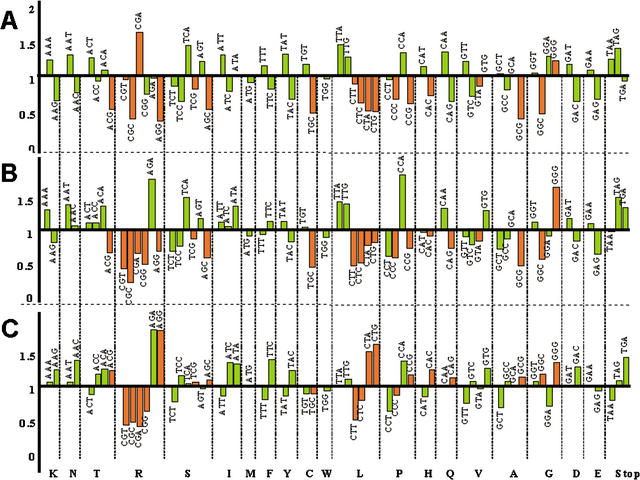
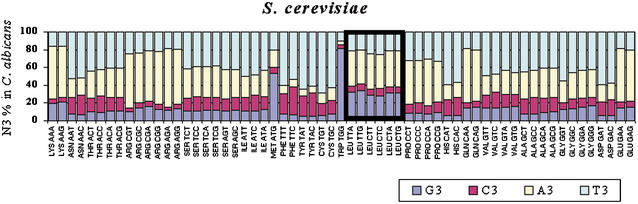
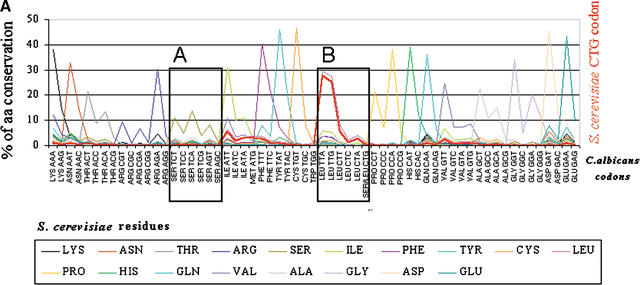
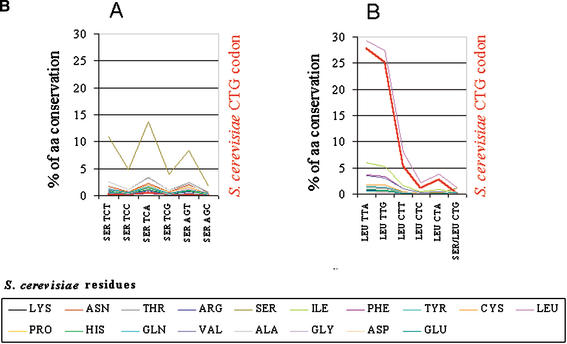
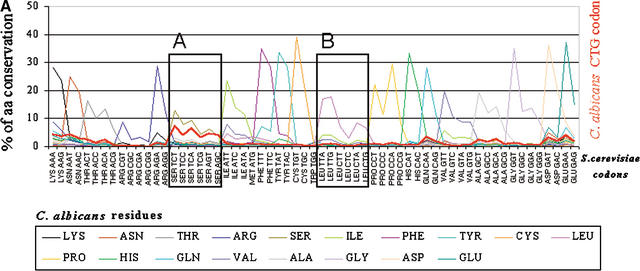
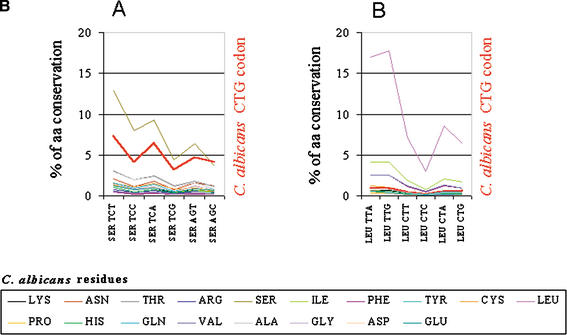
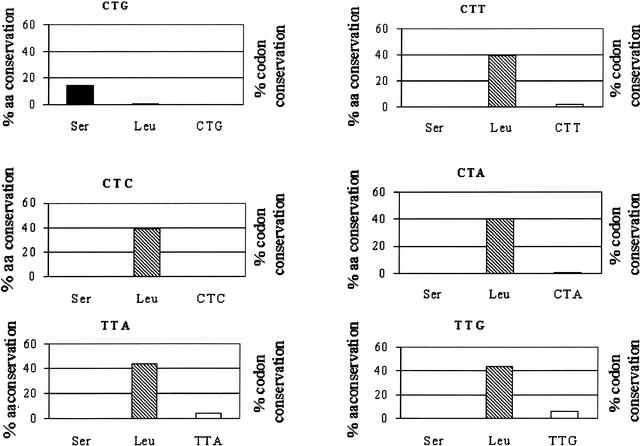
Using the (near) complete genome sequences of the yeasts Candida albicans, Saccharomyces cerevisiae, and Schizosaccharomyces pombe, we address the evolution of a unique genetic code change, which involves decoding of the standard leucine-CTG codon as serine in Candida spp. By using two complementary comparative genomics approaches, we have been able to shed new light on both the origin of the novel Candida spp. Ser-tRNA(CAG), which has mediated CTG reassignment, and on the evolution of the CTG codon in the genomes of C. albicans, S. cerevisiae, and S. pombe. Sequence analyses of newly identified tRNAs from the C. albicans genome demonstrate that the Ser-tRNA(CAG) is derived from a serine and not a leucine tRNA in the ancestor yeast species and that this codon reassignment occurred approximately 170 million years ago, but the origin of the Ser-tRNA(CAG) is more ancient, implying that the ancestral Leu-tRNA that decoded the CTG codon was lost after the appearance of the Ser-tRNA(CAG). Ambiguous CTG decoding by the Ser-tRNA(CAG) combined with biased AT pressure forced the evolution of CTG into TTR codons and have been major forces driving evolution of the CTN codon family in C. albicans. Remarkably, most of the CTG codons present in extant C. albicans genes are encoded by serine and not leucine codons in homologous S. cerevisiae and S. pombe genes, indicating that a significant number of serine TCN and AGY codons evolved into CTG codons either directly by simultaneous double mutations or indirectly through an intermediary codon. In either case, CTG reassignment had a major impact on the evolution of the coding component of the Candida spp. genome.
Figures
References
-
- Averof M., Rokas, A., Wolfe, K.H., and Sharp, P.M. 2000. Evidence for a high frequency of simultaneous double-nucleotide substitutions. Science 287: 1283-1286. - PubMed
-
- Bjork G.R. 1986. Transfer RNA modification in different organisms. Chem. Scripta 26: 91-95.
-
- Bjork G.R., Ericson, J.U., Gustafsson, C.E., Hagervall, T.G., Jonsson, Y.H., and Wikstrom, P.M. 1987. Transfer RNA modification. Annu. Rev. Biochem. 56: 263-287. - PubMed
-
- Bjork G.R., Wikstrom, P.M., and Bystrom, A.S. 1989. Prevention of translational frameshifting by the modified nucleoside 1-methylguanosine. Science 244: 986-989. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous