

Ran's C-terminal, basic patch, and nucleotide exchange mechanisms in light of a canonical structure for Rab, Rho, Ras, and Ran GTPases
- PMID: 12671004
- PMCID: PMC430177
- DOI: 10.1101/gr.862303
Ran's C-terminal, basic patch, and nucleotide exchange mechanisms in light of a canonical structure for Rab, Rho, Ras, and Ran GTPases
Abstract
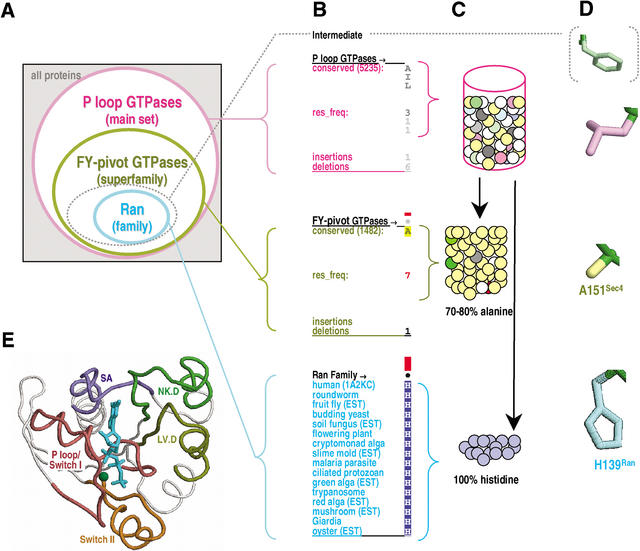
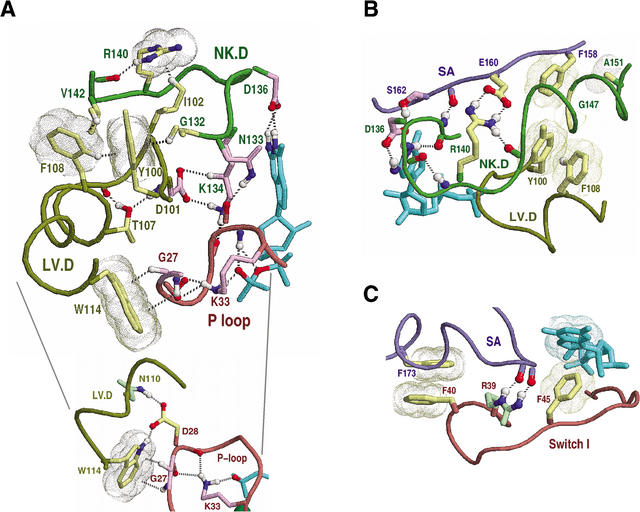
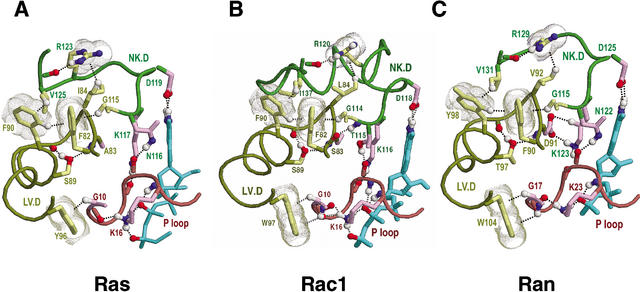
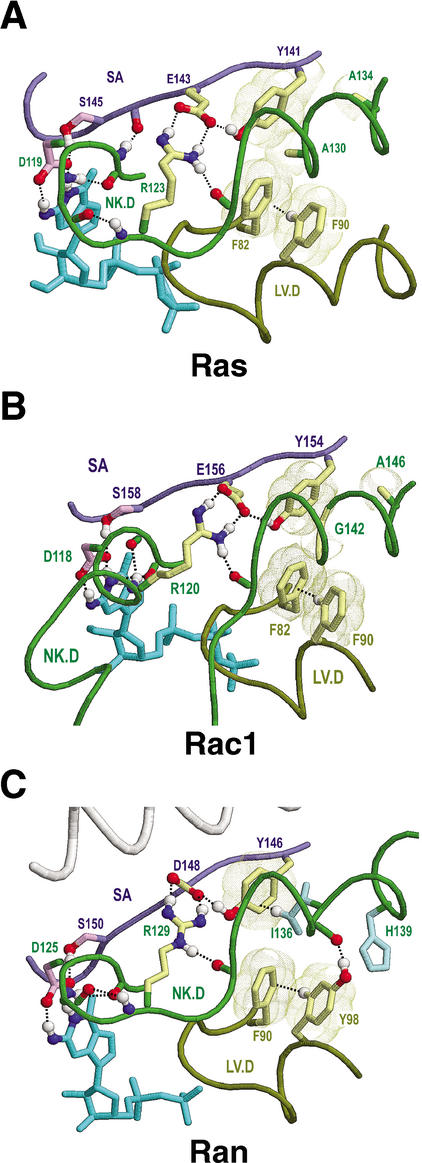
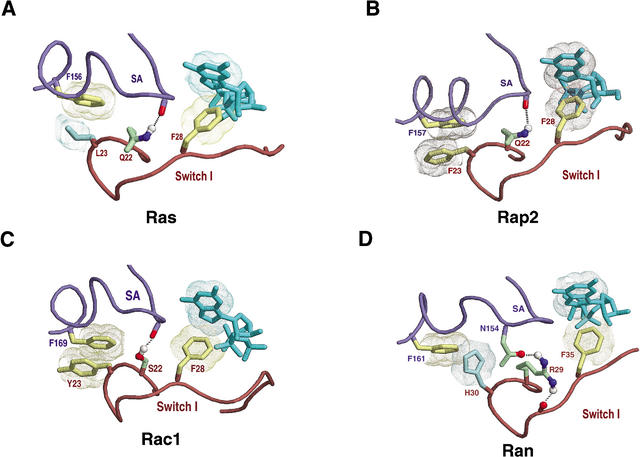
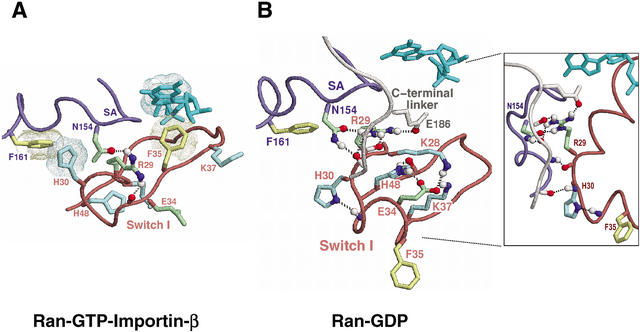
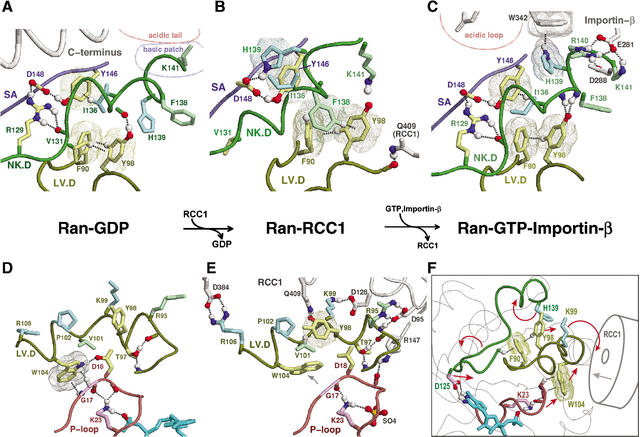
Proteins comprising the core of the eukaryotic cellular machinery are often highly conserved, presumably due to selective constraints maintaining important structural features. We have developed statistical procedures to decompose these constraints into distinct categories and to pinpoint critical structural features within each category. When applied to P-loop GTPases, this revealed within Rab, Rho, Ras, and Ran a canonical network of molecular interactions centered on bound nucleotide. This network presumably performs a crucial structural and/or mechanistic role considering that it has persisted for more than a billion years after the divergence of these families. We call these 'FY-pivot' GTPases after their most distinguishing feature, a phenylalanine or tyrosine that functions as a pivot within this network. Specific families deviate somewhat from canonical features in interesting ways, presumably reflecting their functional specialization during evolution. We illustrate this here for Ran GTPases, within which two highly conserved histidines, His30 and His139, strikingly diverge from their canonical counterparts. These, along with other residues specifically conserved in Ran, such as Tyr98, Lys99, and Phe138, appear to work in conjunction with FY-pivot canonical residues to facilitate alternative conformations in which these histidines are strategically positioned to couple Ran's basic patch and C-terminal switch to nucleotide exchange and effector binding. Other core components of the cellular machinery are likewise amenable to this approach, which we term Contrast Hierarchical Alignment and Interaction Network (CHAIN) analysis.
Figures



References
-
- Alberts B. 1998. The cell as a collection of protein machines: Preparing the next generation of molecular biologists. Cell 92: 291-294. - PubMed
-
- Andrade M.A. and Bork, P. 1995. HEAT repeats in the Huntington's disease protein. Nat. Genet. 11: 115-116. - PubMed
-
- Baker E.N. and Hubbard, R.E. 1984. Hydrogen bonding in globular proteins. Prog. Biophys. Mol. Biol. 44: 97-179. - PubMed
-
- Bar-Sagi D. and Hall, A. 2000. Ras and Rho GTPases: A family reunion. Cell 103: 227-238. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous