

The microRNAs of Caenorhabditis elegans
- PMID: 12672692
- PMCID: PMC196042
- DOI: 10.1101/gad.1074403
The microRNAs of Caenorhabditis elegans
Abstract
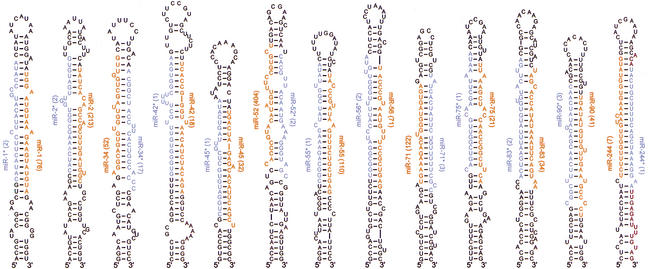
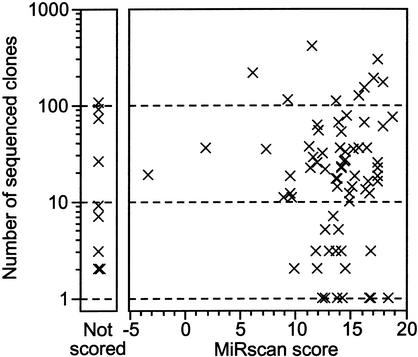
MicroRNAs (miRNAs) are an abundant class of tiny RNAs thought to regulate the expression of protein-coding genes in plants and animals. In the present study, we describe a computational procedure to identify miRNA genes conserved in more than one genome. Applying this program, known as MiRscan, together with molecular identification and validation methods, we have identified most of the miRNA genes in the nematode Caenorhabditis elegans. The total number of validated miRNA genes stands at 88, with no more than 35 genes remaining to be detected or validated. These 88 miRNA genes represent 48 gene families; 46 of these families (comprising 86 of the 88 genes) are conserved in Caenorhabditis briggsae, and 22 families are conserved in humans. More than a third of the worm miRNAs, including newly identified members of the lin-4 and let-7 gene families, are differentially expressed during larval development, suggesting a role for these miRNAs in mediating larval developmental transitions. Most are present at very high steady-state levels-more than 1000 molecules per cell, with some exceeding 50,000 molecules per cell. Our census of the worm miRNAs and their expression patterns helps define this class of noncoding RNAs, lays the groundwork for functional studies, and provides the tools for more comprehensive analyses of miRNA genes in other species.
Figures
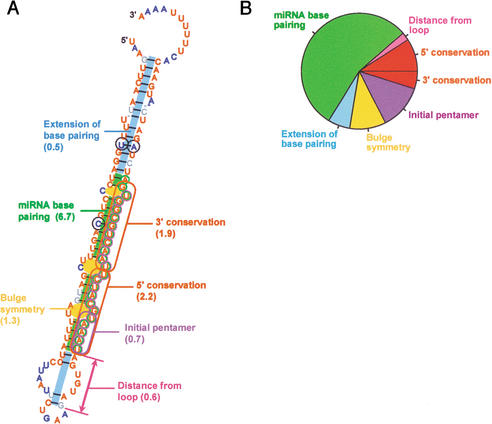
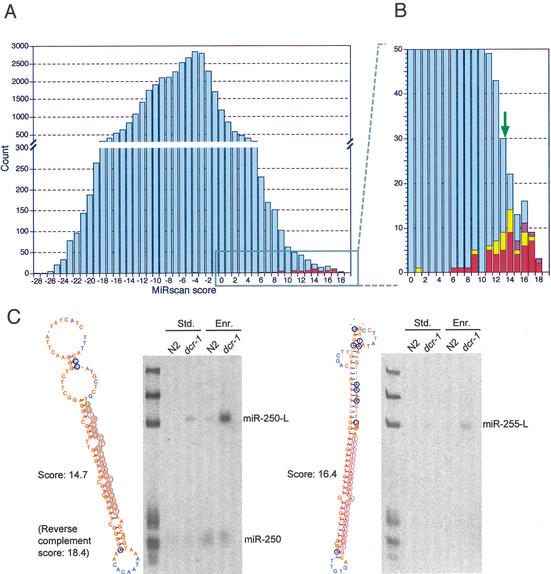

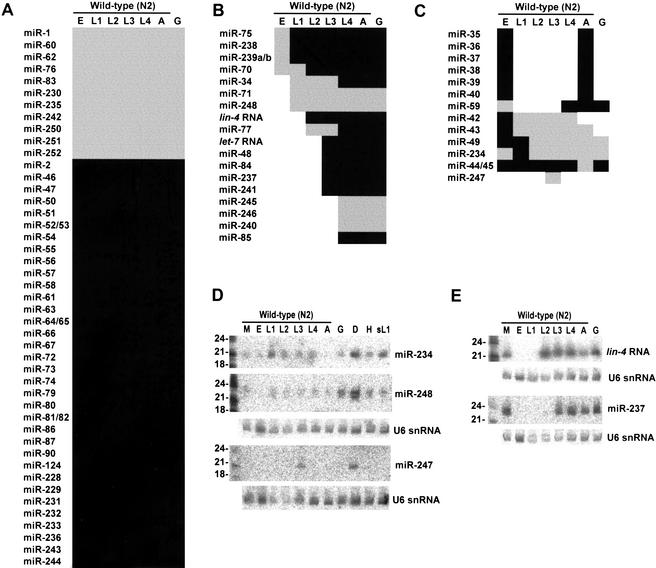
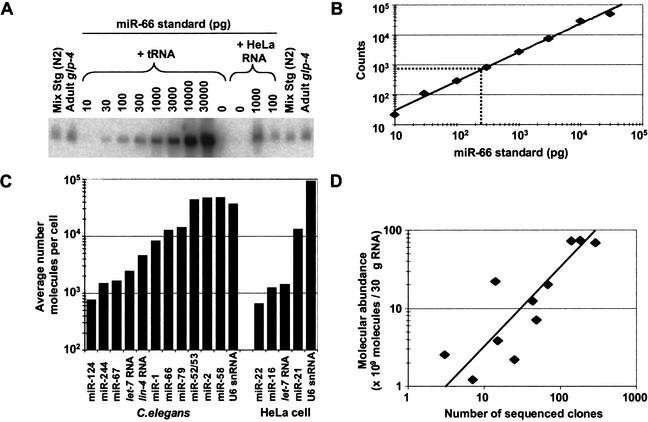
References
-
- Aravin AA, Naumova NM, Tulin AA, Rozovsky YM, Gvozdev VA. Double-stranded RNA-mediated silencing of genomic tandem repeats and transposable elements in D. melanogaster germline. Curr Biol. 2001;11:1017–1027. - PubMed
-
- Argaman L, Hershberg R, Vogel J, Bejerano G, Wagner EG, Margalit H, Altuvia S. Novel small RNA-encoding genes in the intergenic regions of Escherichia coli. Curr Biol. 2001;11:941–950. - PubMed
-
- Beanan MJ, Strome S. Characterization of a germ-line proliferation mutation in C. elegans. Development. 1992;116:755–766. - PubMed
-
- Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001;409:295–296. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases