

Glycogen regulation and functional role in mouse white matter
- PMID: 12679378
- PMCID: PMC2342948
- DOI: 10.1113/jphysiol.2003.042416
Glycogen regulation and functional role in mouse white matter
Abstract
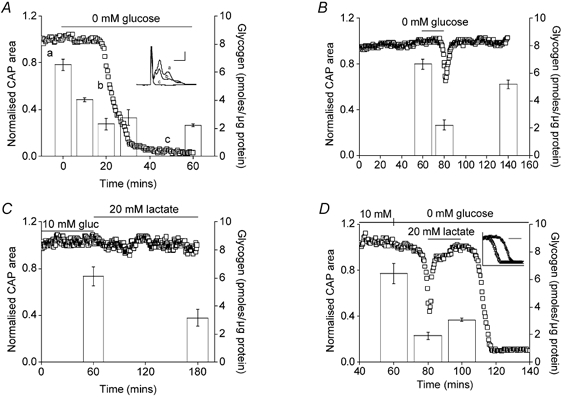
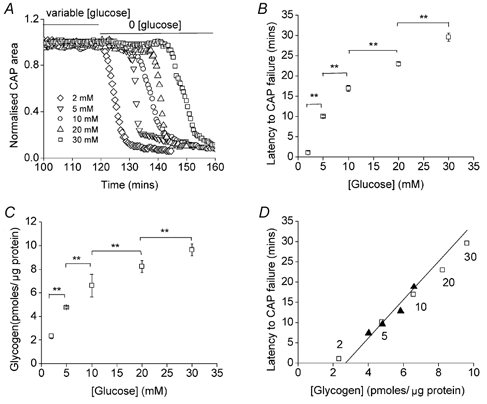
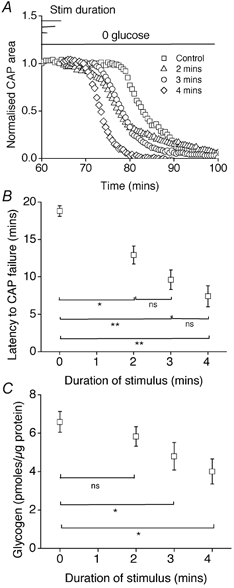
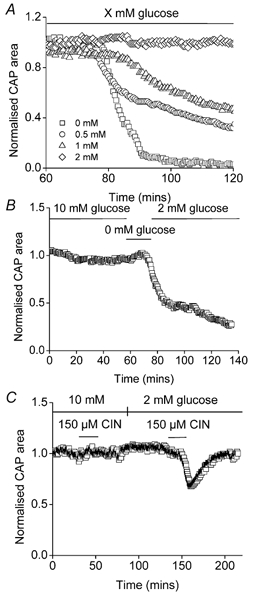
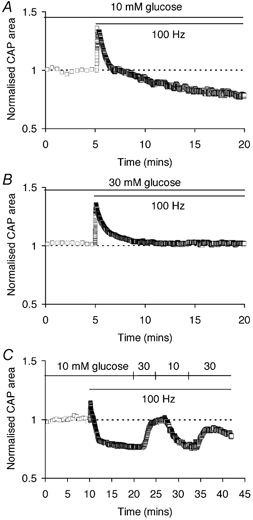
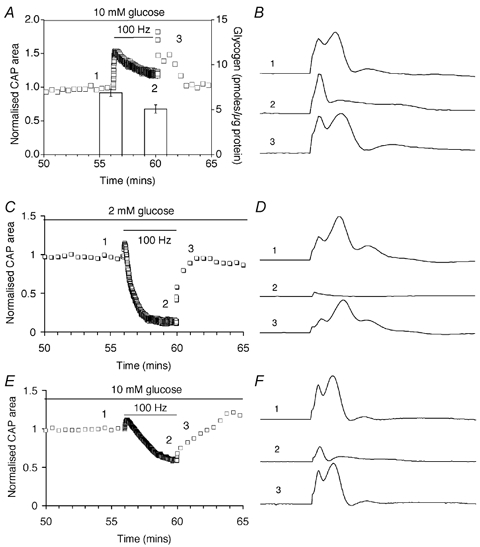
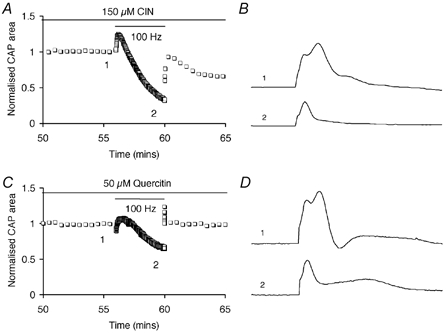
CNS glycogen, contained predominantly in astrocytes, can be converted to a monocarboxylate and transported to axons as an energy source during aglycaemia. We analysed glycogen regulation and the role of glycogen in supporting neural activity in adult mouse optic nerve, a favourable white matter preparation. Axon function was quantified by measuring the compound action potential (CAP) area. During aglycaemia, axon function persisted for 20 min, then declined in conjunction with glycogen content. Lactate fully supported CAPs in the absence of glucose, but was unable to sustain glycogen content; thus, axon failure occurred rapidly when lactate was withdrawn. Glycogen content in the steady state was directly proportional to bath glucose concentration. Increasing [K+]o to 10 mM caused a rapid decrease in glycogen content. Latency to onset of CAP failure during aglycaemia was directly proportional to glycogen content and varied from about 2 to 30 min. Intense neural activity reduced glycogen content in the presence of 10 mM bath glucose and CAP area gradually declined. CAP area declined more rapidly during high frequency stimulation if monocarboxylate transport was inhibited. This suggested that astrocytic glycogen was broken down to a monocarboxylate(s) that was used by rapidly discharging axons. Likewise, depleting glycogen by brief periods of high frequency axon stimulation accelerated onset of CAP decline during aglycaemia. In summary, these experiments indicated that glycogen content was under dynamic control and that glycogen was used to support the energy needs of CNS axons during both physiological as well as pathological processes.
Figures
References
-
- Baltan Tekkök S, Brown AM, Ransom BR. Persistence of axonal function during anoxic insult in mouse optic nerve. Glia. 2002;38(suppl. 1):245.
-
- Belt JA, Thomas JA, Buchsbaum RN, Racker E. Inhibition of lactate transport and glycolysis in Ehrlich ascites tumor cells by bioflavonoids. Biochemistry. 1979;18:3506–3511. - PubMed
-
- Brown AM. A step-by-step guide to non-linear regression analysis of experimental data using a Microsoft Excel spreadsheet. Comp Meth Prog Biomed. 2001;65:191–200. - PubMed
-
- Brown AM, Ransom BR. Astrocytic glycogen maintains axon function during periods of hypoglycaemia in central white matter. JPhysiol. 2001;536.P:118P.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous