

Genome sequence of Chlamydophila caviae (Chlamydia psittaci GPIC): examining the role of niche-specific genes in the evolution of the Chlamydiaceae
- PMID: 12682364
- PMCID: PMC153749
- DOI: 10.1093/nar/gkg321
Genome sequence of Chlamydophila caviae (Chlamydia psittaci GPIC): examining the role of niche-specific genes in the evolution of the Chlamydiaceae
Abstract
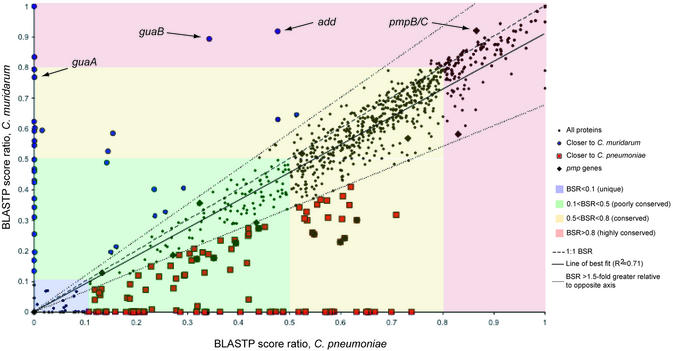
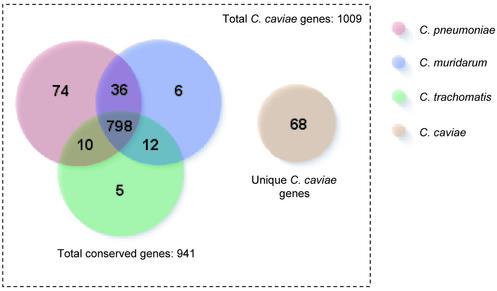
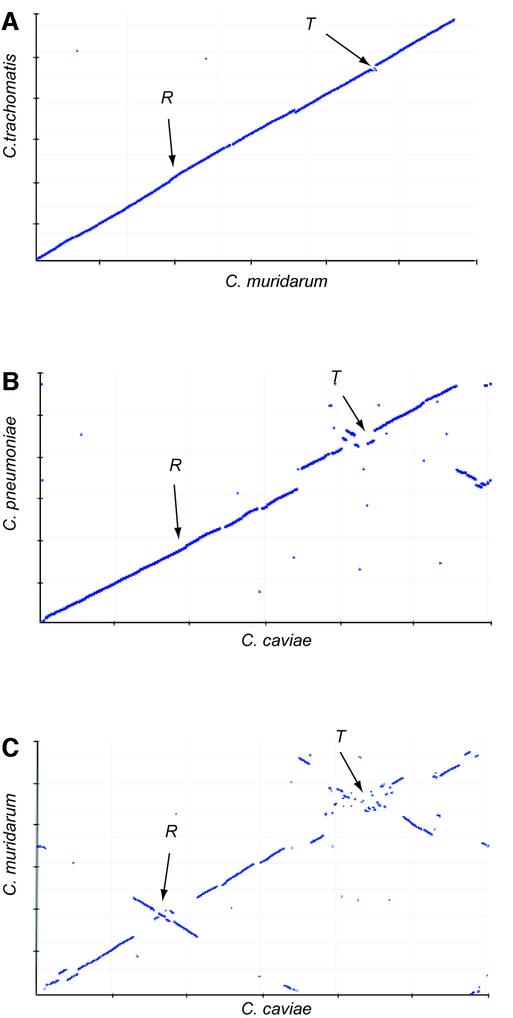
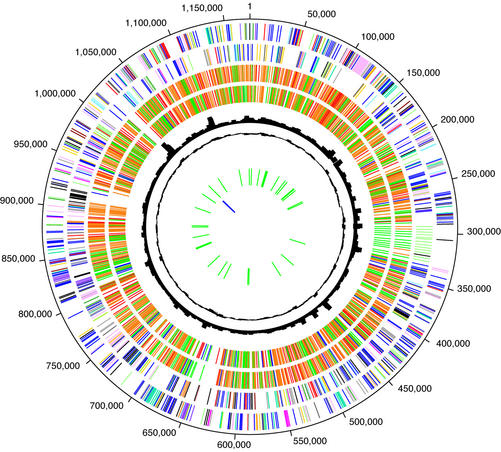
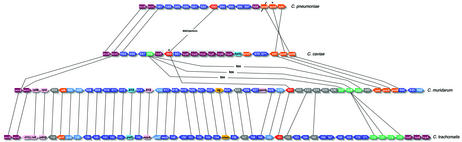
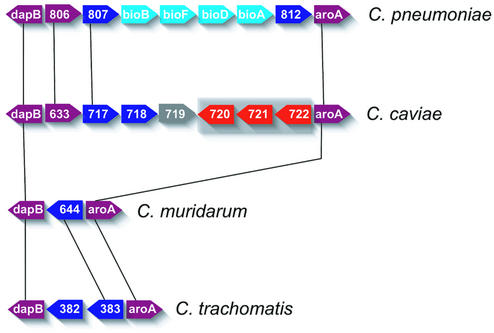
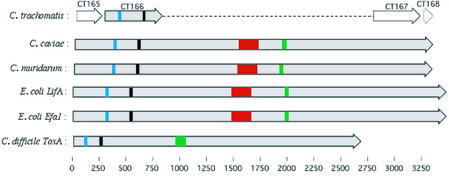
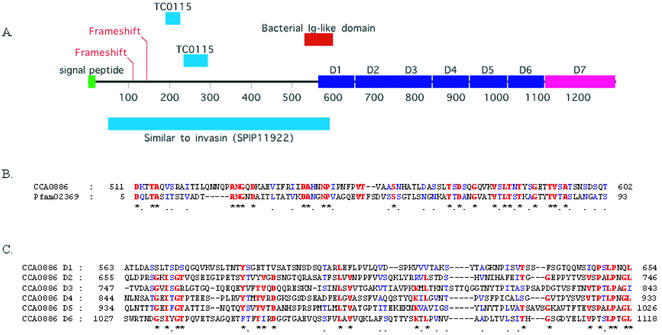
The genome of Chlamydophila caviae (formerly Chlamydia psittaci, GPIC isolate) (1 173 390 nt with a plasmid of 7966 nt) was determined, representing the fourth species with a complete genome sequence from the Chlamydiaceae family of obligate intracellular bacterial pathogens. Of 1009 annotated genes, 798 were conserved in all three other completed Chlamydiaceae genomes. The C.caviae genome contains 68 genes that lack orthologs in any other completed chlamydial genomes, including tryptophan and thiamine biosynthesis determinants and a ribose-phosphate pyrophosphokinase, the product of the prsA gene. Notable amongst these was a novel member of the virulence-associated invasin/intimin family (IIF) of Gram-negative bacteria. Intriguingly, two authentic frameshift mutations in the ORF indicate that this gene is not functional. Many of the unique genes are found in the replication termination region (RTR or plasticity zone), an area of frequent symmetrical inversion events around the replication terminus shown to be a hotspot for genome variation in previous genome sequencing studies. In C.caviae, the RTR includes several loci of particular interest including a large toxin gene and evidence of ancestral insertion(s) of a bacteriophage. This toxin gene, not present in Chlamydia pneumoniae, is a member of the YopT effector family of type III-secreted cysteine proteases. One gene cluster (guaBA-add) in the RTR is much more similar to orthologs in Chlamydia muridarum than those in the phylogenetically closest species C.pneumoniae, suggesting the possibility of horizontal transfer of genes between the rodent-associated Chlamydiae. With most genes observed in the other chlamydial genomes represented, C.caviae provides a good model for the Chlamydiaceae and a point of comparison against the human atherosclerosis-associated C.pneumoniae. This crucial addition to the set of completed Chlamydiaceae genome sequences is enabling dissection of the roles played by niche-specific genes in these important bacterial pathogens.
Figures
References
-
- Everett K.D., Bush,R.M. and Andersen,A.A. (1999) Emended description of the order Chlamydiales, proposal of Parachlamydiaceae fam. nov. and Simkaniaceae fam. nov., each containing one monotypic genus, revised taxonomy of the family Chlamydiaceae, including a new genus and five new species and standards for the identification of organisms. Int. J. Syst. Bacteriol., 49, 415–440. - PubMed
-
- Schachter J. (1999) Infection and disease epidemiology. In Stephens,R.S. (ed.), Chlamydia: Intracellular Biology, Pathogenesis and Immunity. ASM Press, Washington, DC, pp. 139–169.
-
- Grayston J.T. (1992) Chlamydia pneumoniae, strain TWAR pneumonia. Annu. Rev. Med., 43, 317–323. - PubMed
-
- Hahn D.L., Azenabor,A.A., Beatty,W.L. and Byrne,G.I. (2002). Chlamydia pneumoniae as a respiratory pathogen. Front Biosci., 7, e66–e76. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
