

Erythropoietin fosters both intrinsic and extrinsic neuronal protection through modulation of microglia, Akt1, Bad, and caspase-mediated pathways
- PMID: 12684267
- PMCID: PMC1573758
- DOI: 10.1038/sj.bjp.0705161
Erythropoietin fosters both intrinsic and extrinsic neuronal protection through modulation of microglia, Akt1, Bad, and caspase-mediated pathways
Abstract
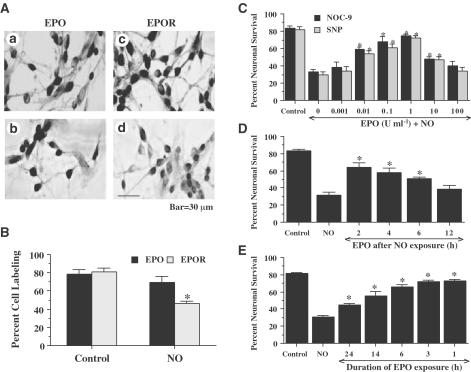
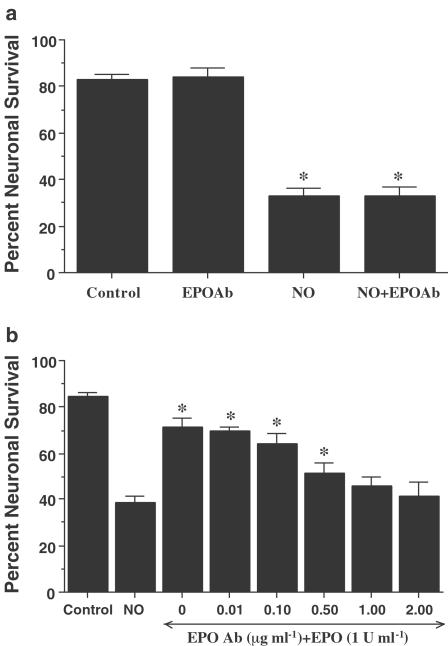
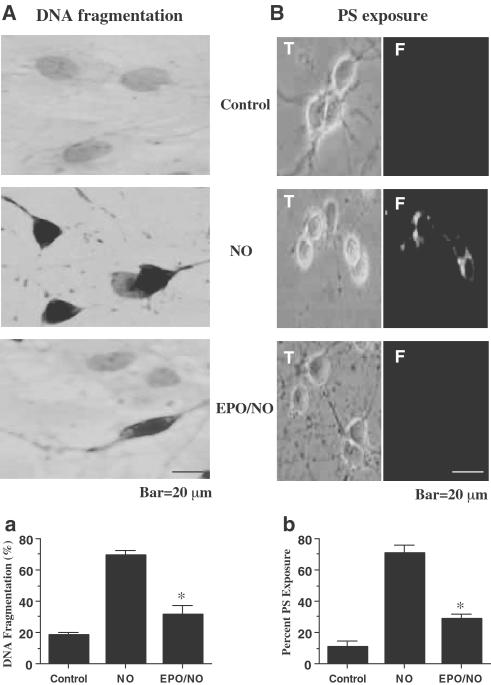
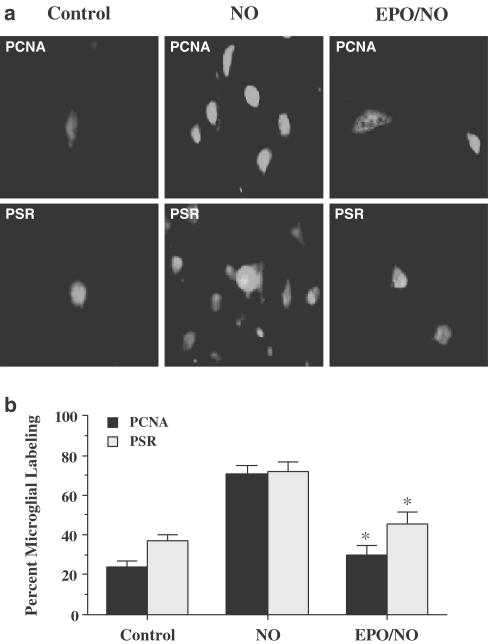
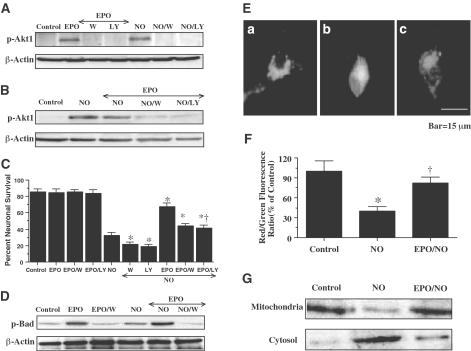
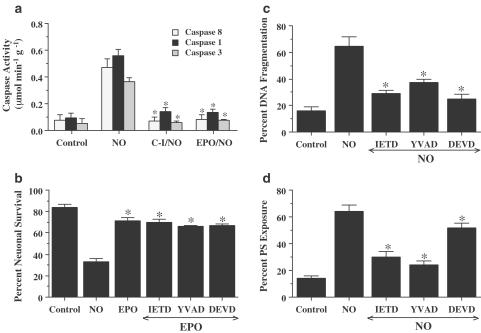
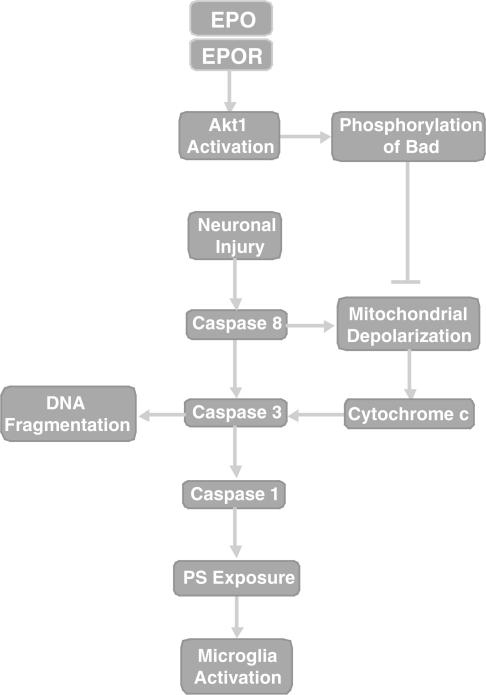
1. Erythropoietin (EPO) plays a significant role in the hematopoietic system, but the function of EPO as a neuroprotectant and anti-inflammatory mediator requires further definition. We therefore examined the cellular mechanisms that mediate protection by EPO during free radical injury in primary neurons and cerebral microglia. 2. Neuronal injury was evaluated by trypan blue, DNA fragmentation, phosphatidylserine (PS) exposure, Akt1 phosphorylation, Bad phosphorylation, mitochondrial membrane potential, and cysteine protease activity. Microglial activation was assessed through proliferating cell nuclear antigen and PS receptor expression. 3. EPO provides intrinsic neuronal protection that is both necessary and sufficient to prevent acute genomic DNA destruction and subsequent membrane PS exposure, since protection by EPO is completely abolished by cotreatment with an anti-EPO neutralizing antibody. 4. Extrinsic protection by EPO is offered through the inhibition of cerebral microglial activation and the suppression of microglial PS receptor expression for the prevention of neuronal phagocytosis. In regards to microglial chemotaxis, EPO modulates neuronal poptotic membrane PS exposure necessary for microglial activation primarily through the regulation of caspase 1. 5. EPO increases Akt1 activity, phosphorylates Bad, and maintains neuronal nuclear DNA integrity through the downstream modulation of mitochrondrial membrane potential, cytochrome c release, and caspase 1, 3, and 8-like activities. 6. Elucidating the intrinsic and extrinsic protective pathways of EPO that mediate both neuronal integrity and inflammatory microglial activation may enhance the development of future therapies directed against acute neuronal injury.
Figures
References
-
- ACS G., ACS P., BECKWITH S.M., PITTS R.L., CLEMENTS E., WONG K., VERMA A. Erythropoietin and erythropoietin receptor expression in human cancer. Cancer Res. 2001;61:3561–3565. - PubMed
-
- ANDERSON I., ADINOLFI C., DOCTROW S., HUFFMAN K., JOY K.A., MALFROY B., SODEN P., RUPNIAK H.T., BARNES J.C. Oxidative signalling and inflammatory pathways in Alzheimer's disease. Biochem. Soc. Symp. 2001;67:141–149. - PubMed
-
- BAL-PRICE A., BROWN G.C. Nitric-oxide-induced necrosis and apoptosis in PC12 cells mediated by mitochondria. J. Neurochem. 2000;75:1455–1464. - PubMed
-
- BERNAUDIN M., MARTI H.H., ROUSSEL S., DIVOUX D., NOUVELOT A., MACKENZIE E.T., PETIT E. A potential role for erythropoietin in focal permanent cerebral ischemia in mice. J. Cerer. Blood Flow Metab. 1999;19:643–651. - PubMed
-
- BLUME-JENSEN P., JANKNECHT R., HUNTER T. The kit receptor promotes cell survival via activation of PI 3-kinase and subsequent Akt-mediated phosphorylation of Bad on Ser136. Curr. Biol. 1998;8:779–782. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
