

Simulating disorder-order transitions in molecular recognition of unstructured proteins: where folding meets binding
- PMID: 12697905
- PMCID: PMC154313
- DOI: 10.1073/pnas.0531373100
Simulating disorder-order transitions in molecular recognition of unstructured proteins: where folding meets binding
Abstract
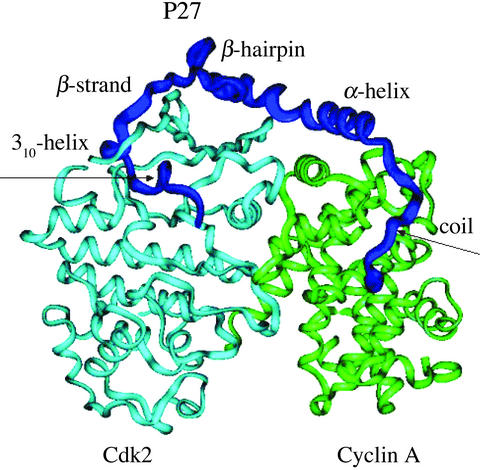
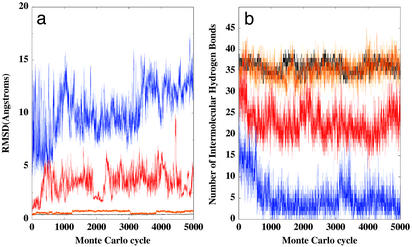
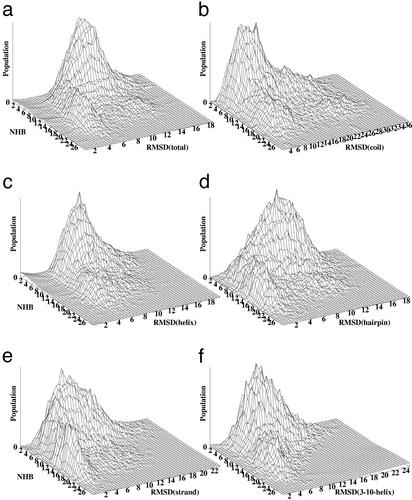
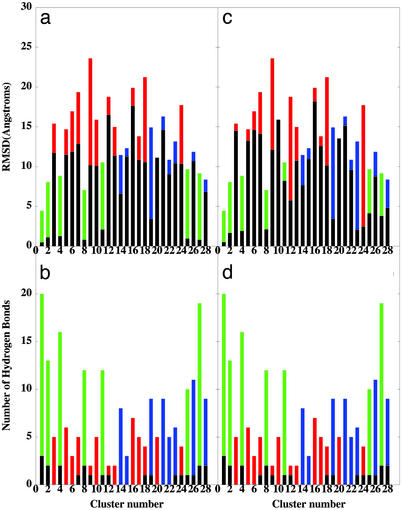
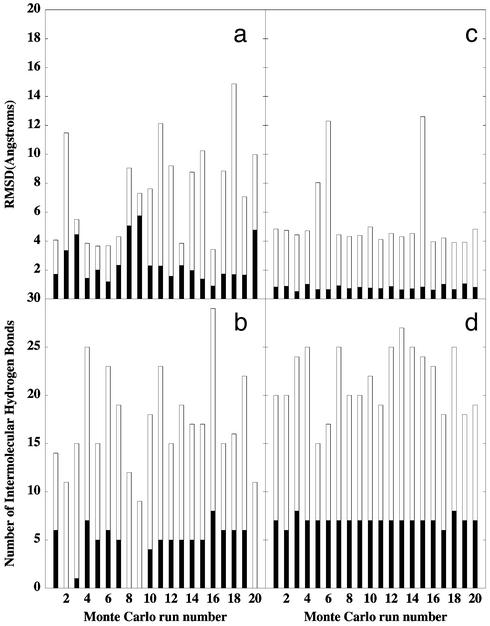
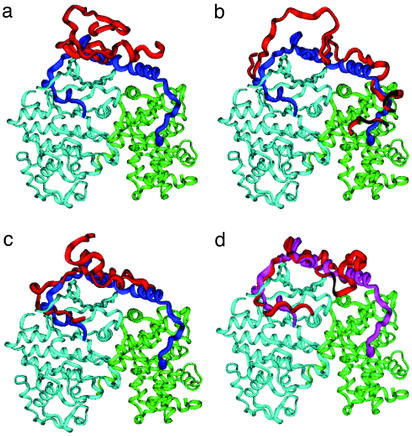
A microscopic study of functional disorder-order folding transitions coupled to binding is performed for the p27 protein, which derives a kinetic advantage from the intrinsically disordered unbound form on binding with the phosphorylated cyclin A-cyclin-dependent kinase 2 (Cdk2) complex. Hierarchy of structural loss during p27 coupled unfolding and unbinding is simulated by using high-temperature Monte Carlo simulations initiated from the crystal structure of the tertiary complex. Subsequent determination of the transition-state ensemble and the proposed atomic picture of the folding mechanism coupled to binding provide a microscopic rationale that reconciles the initiation recruitment of p27 at the cyclin A docking site with the kinetic benefit for a disordered alpha-helix in the unbound form of p27. The emerging structural polarization in the ensemble of unfolding/unbinding trajectories and in the computationally determined transition-state ensemble is not determined by the intrinsic folding preferences of p27 but rather is attributed to the topological requirements of the native intermolecular interface to order beta-hairpin and beta-strand of p27 that could be critical for nucleating rapid folding transition coupled to binding. In agreement with the experimental data, the disorder-order folding transition for p27 is largely determined by the functional requirement to form a specific intermolecular interface that ultimately dictates the folding mechanism and overwhelms any local folding preferences for creating a stable alpha-helix in the p27 structure before overcoming the major free energy barrier.
Figures
Similar articles
-
Protein conformational transitions coupled to binding in molecular recognition of unstructured proteins: deciphering the effect of intermolecular interactions on computational structure prediction of the p27Kip1 protein bound to the cyclin A-cyclin-dependent kinase 2 complex.Proteins. 2005 Feb 15;58(3):706-16. doi: 10.1002/prot.20351. Proteins. 2005. PMID: 15609350
-
Protein conformational transitions coupled to binding in molecular recognition of unstructured proteins: hierarchy of structural loss from all-atom Monte Carlo simulations of p27Kip1 unfolding-unbinding and structural determinants of the binding mechanism.Biopolymers. 2004 Dec 5;75(5):420-33. doi: 10.1002/bip.20149. Biopolymers. 2004. PMID: 15468065
-
Topology-based modeling of intrinsically disordered proteins: balancing intrinsic folding and intermolecular interactions.Proteins. 2011 Apr;79(4):1251-66. doi: 10.1002/prot.22960. Epub 2011 Jan 25. Proteins. 2011. PMID: 21268115
-
Protein modeling with reduced representation: statistical potentials and protein folding mechanism.Acta Biochim Pol. 2005;52(4):741-8. Epub 2005 May 31. Acta Biochim Pol. 2005. PMID: 15933762 Review.
-
Limited cooperativity in protein folding.Curr Opin Struct Biol. 2016 Feb;36:58-66. doi: 10.1016/j.sbi.2015.12.001. Epub 2016 Feb 2. Curr Opin Struct Biol. 2016. PMID: 26845039 Review.
Cited by
-
Characterization of the structure and intermolecular interactions between the connexin 32 carboxyl-terminal domain and the protein partners synapse-associated protein 97 and calmodulin.J Biol Chem. 2012 Aug 10;287(33):27771-88. doi: 10.1074/jbc.M112.382572. Epub 2012 Jun 20. J Biol Chem. 2012. PMID: 22718765 Free PMC article.
-
Residual structures, conformational fluctuations, and electrostatic interactions in the synergistic folding of two intrinsically disordered proteins.PLoS Comput Biol. 2012 Jan;8(1):e1002353. doi: 10.1371/journal.pcbi.1002353. Epub 2012 Jan 12. PLoS Comput Biol. 2012. PMID: 22253588 Free PMC article.
-
Allosteric modulators of steroid hormone receptors: structural dynamics and gene regulation.Endocr Rev. 2012 Apr;33(2):271-99. doi: 10.1210/er.2011-1033. Epub 2012 Mar 20. Endocr Rev. 2012. PMID: 22433123 Free PMC article. Review.
-
Dentin Sialophophoprotein (DSPP) and Dentin.J Oral Biosci. 2008 Jan 1;50(1):33-44. doi: 10.2330/joralbiosci.50.33. J Oral Biosci. 2008. PMID: 20037676 Free PMC article.
-
Intrinsically disordered proteins in a physics-based world.Int J Mol Sci. 2010;11(12):5292-309. doi: 10.3390/ijms11125292. Epub 2010 Dec 21. Int J Mol Sci. 2010. PMID: 21614208 Free PMC article. Review.
References
-
- Wright P E, Dyson H J. J Mol Biol. 1999;293:321–331. - PubMed
-
- Dunker A K, Lawson J D, Brown C J, Williams R M, Romero P, Oh J S, Oldfield C J, Campen A M, Obradovic Z. J Mol Graphics Model. 2001;19:26–59. - PubMed
-
- Dyson H J, Wright P E. Curr Opin Struct Biol. 2002;12:54–60. - PubMed
-
- Spolar R S, Record M T. Science. 1994;263:777–784. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
