

Production of MPS VII mouse (Gus(tm(hE540A x mE536A)Sly)) doubly tolerant to human and mouse beta-glucuronidase
- PMID: 12700165
- PMCID: PMC1567498
- DOI: 10.1093/hmg/ddg119
Production of MPS VII mouse (Gus(tm(hE540A x mE536A)Sly)) doubly tolerant to human and mouse beta-glucuronidase
Abstract
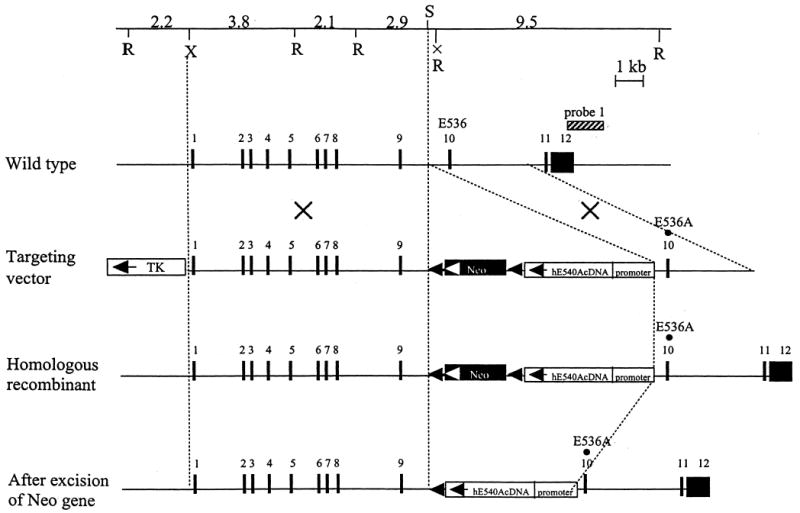
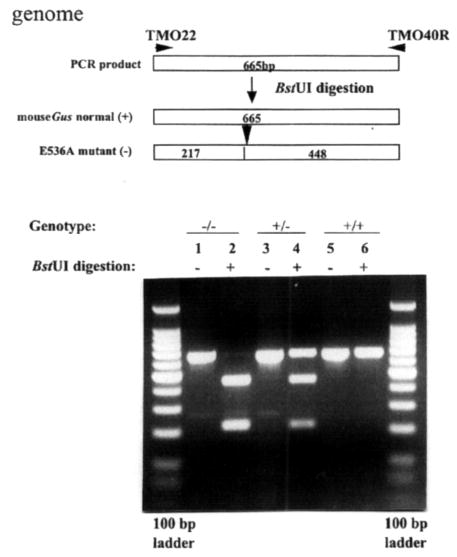
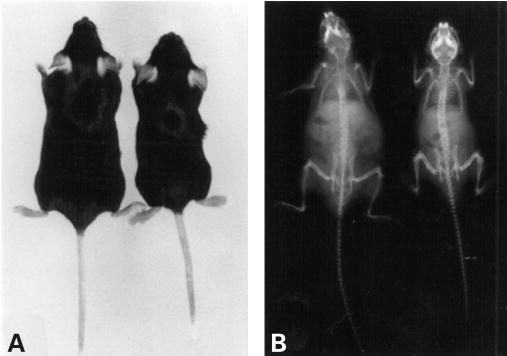
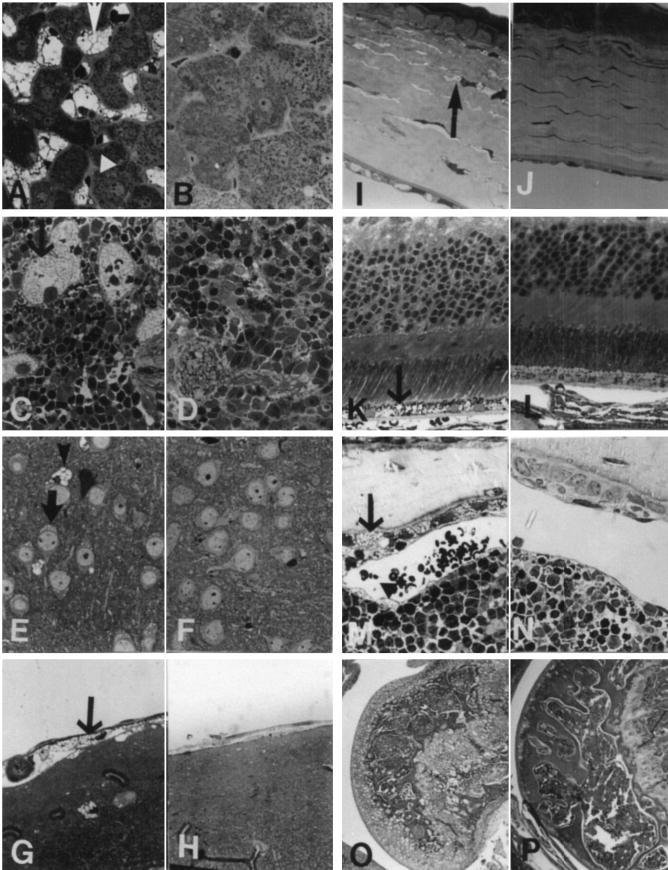
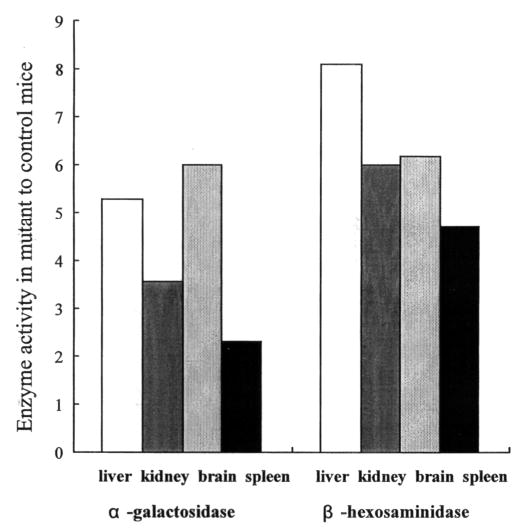
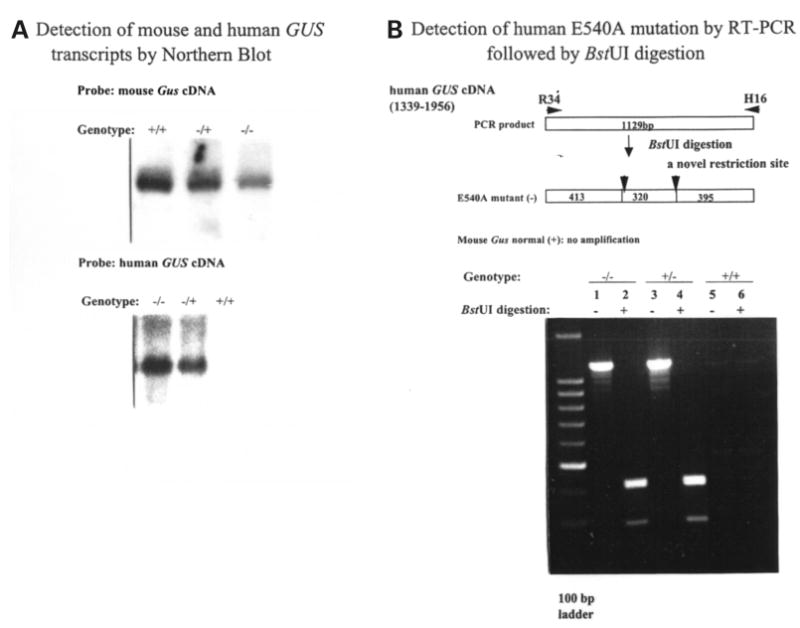
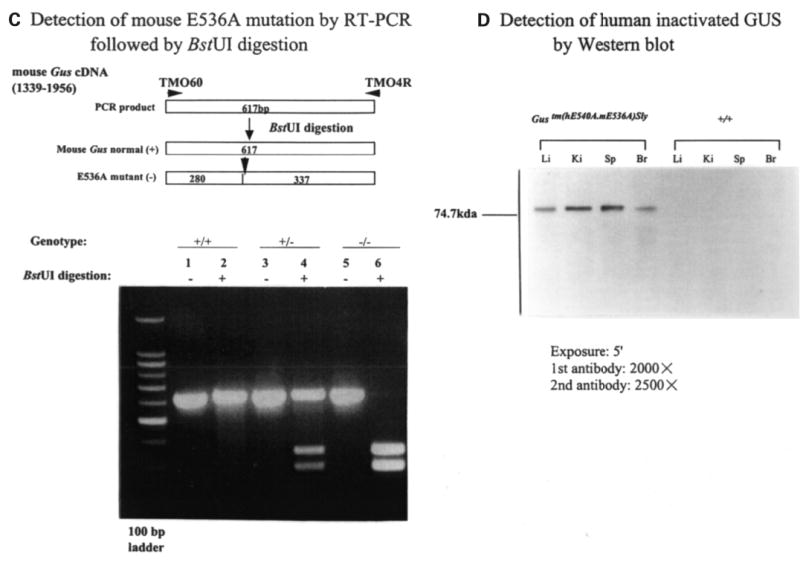
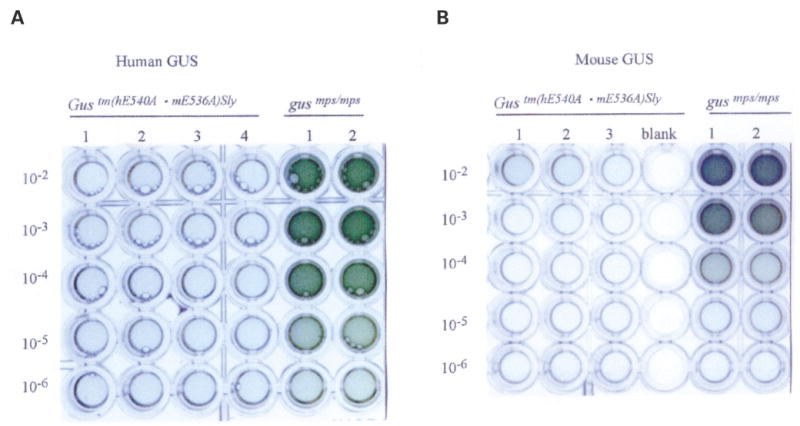
Mucopolysaccharidosis VII (MPS VII, Sly syndrome) is an autosomal recessive lysosomal storage disease caused by beta-glucuronidase (GUS) deficiency. A naturally occurring mouse model of that disease has been very useful for studying experimental approaches to therapy. However, immune responses can complicate evaluation of the long-term benefits of enzyme replacement or gene therapy delivered to adult MPS VII mice. To make this model useful for studying the long-term effectiveness and side effects of experimental therapies delivered to adult mice, we developed a new MPS VII mouse model, which is tolerant to both human and murine GUS. To achieve this, we used homologous recombination to introduce simultaneously a human cDNA transgene expressing inactive human GUS into intron 9 of the murine Gus gene and a targeted active site mutation (E536A) into the adjacent exon 10. When the heterozygote products of germline transmission were bred to homozygosity, the homozygous mice expressed no GUS enzyme activity but expressed inactive human GUS protein highly and were tolerant to immune challenge with human enzyme. Expression of the mutant murine Gus gene was reduced to about 10% of normal levels, but the inactive murine GUS enzyme also conferred tolerance to murine GUS. This MPS VII mouse model should be useful to evaluate therapeutic responses in adult mice receiving repetitive doses of enzyme or mice receiving gene therapy as adults. Heterozygotes expressed only 9.5-26% of wild-type levels of murine GUS instead of the expected 50%, indicating a dominant-negative effect of the mutant enzyme monomers on the activity of GUS tetramers in different tissues. Corrective gene therapy in this model should provide high enough levels of expression of normal GUS monomers to overcome the dominant negative effect of mutant monomers on newly synthesized GUS tetramers in most tissues.
Figures
Similar articles
-
Mutations and polymorphisms in GUSB gene in mucopolysaccharidosis VII (Sly Syndrome).Hum Mutat. 2009 Apr;30(4):511-9. doi: 10.1002/humu.20828. Hum Mutat. 2009. PMID: 19224584 Free PMC article. Review.
-
Active site mutant transgene confers tolerance to human beta-glucuronidase without affecting the phenotype of MPS VII mice.Proc Natl Acad Sci U S A. 2001 Feb 27;98(5):2205-10. doi: 10.1073/pnas.051623698. Epub 2001 Feb 13. Proc Natl Acad Sci U S A. 2001. PMID: 11226217 Free PMC article.
-
Missense models [Gustm(E536A)Sly, Gustm(E536Q)Sly, and Gustm(L175F)Sly] of murine mucopolysaccharidosis type VII produced by targeted mutagenesis.Proc Natl Acad Sci U S A. 2002 Nov 12;99(23):14982-7. doi: 10.1073/pnas.232570999. Epub 2002 Oct 28. Proc Natl Acad Sci U S A. 2002. PMID: 12403825 Free PMC article.
-
Lentiviral-mediated gene therapy results in sustained expression of β-glucuronidase for up to 12 months in the gus(mps/mps) and up to 18 months in the gus(tm(L175F)Sly) mouse models of mucopolysaccharidosis type VII.Hum Gene Ther. 2014 Sep;25(9):798-810. doi: 10.1089/hum.2013.141. Epub 2014 Aug 14. Hum Gene Ther. 2014. PMID: 25003807
-
Murine mucopolysaccharidosis type VII: the impact of therapies on the clinical course and pathology in a murine model of lysosomal storage disease.J Inherit Metab Dis. 1998 Aug;21(5):575-86. doi: 10.1023/a:1005423222927. J Inherit Metab Dis. 1998. PMID: 9728337 Review.
Cited by
-
Growth Plate Pathology in the Mucopolysaccharidosis Type VI Rat Model-An Experimental and Computational Approach.Diagnostics (Basel). 2020 May 31;10(6):360. doi: 10.3390/diagnostics10060360. Diagnostics (Basel). 2020. PMID: 32486376 Free PMC article.
-
Polymer-based drug delivery systems under investigation for enzyme replacement and other therapies of lysosomal storage disorders.Adv Drug Deliv Rev. 2023 Jun;197:114683. doi: 10.1016/j.addr.2022.114683. Epub 2023 Jan 16. Adv Drug Deliv Rev. 2023. PMID: 36657645 Free PMC article. Review.
-
Enzyme replacement therapy on hypophosphatasia mouse model.J Inherit Metab Dis. 2014 Mar;37(2):309-317. doi: 10.1007/s10545-013-9646-7. Epub 2013 Aug 27. J Inherit Metab Dis. 2014. PMID: 23978959 Free PMC article.
-
Chronic enzyme replacement therapy ameliorates neuropathology in alpha-mannosidosis mice.Ann Clin Transl Neurol. 2015 Sep 19;2(11):987-1001. doi: 10.1002/acn3.245. eCollection 2015 Nov. Ann Clin Transl Neurol. 2015. PMID: 26817023 Free PMC article.
-
Mutations and polymorphisms in GUSB gene in mucopolysaccharidosis VII (Sly Syndrome).Hum Mutat. 2009 Apr;30(4):511-9. doi: 10.1002/humu.20828. Hum Mutat. 2009. PMID: 19224584 Free PMC article. Review.
References
-
- Sly WS, Quinton BA, McAlister WH, Rimoin DL. Beta-glucuronidase deficiency: report of clinical, radiologic, and biochemical features of a new mucopolysaccharidosis. J Pediatr. 1973;82:249–257. - PubMed
-
- Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. Vol. 3. McGraw-Hill, Medical Publishing Division; New York: 2001. pp. 3421–3452.
-
- Paigen K. Mammalian beta-glucuronidase: genetics, molecular biology, and cell biology. Prog Nucl Acid Res Mol Biol. 1989;37:155–205. - PubMed
-
- Tomatsu S, Sukegawa K, Ikedo Y, Fukuda S, Yamada Y, Sasaki T, Okamoto H, Kuwabara T, Orii T. Molecular basis of mucopolysaccharidosis type VII: replacement of Ala619 in beta-glucuronidase with Val. Gene. 1990;89:283–287. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
