

Review
doi: 10.1128/JB.185.9.2692-2699.2003.
Thirteen years of building constraint-based in silico models of Escherichia coli
Affiliations
- PMID: 12700248
- PMCID: PMC154396
- DOI: 10.1128/JB.185.9.2692-2699.2003
Item in Clipboard
Review
Thirteen years of building constraint-based in silico models of Escherichia coli
J Bacteriol.
2003 May.
No abstract available
Figures
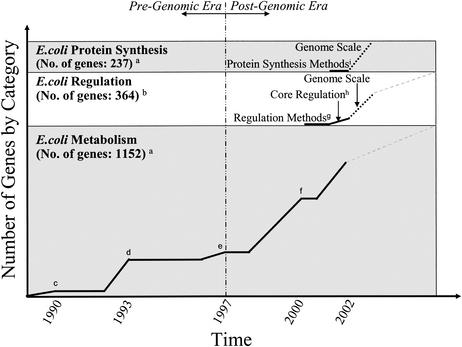
Development of successive constraint-based FBA models of E. coli. Constraint-based models of E. coli first focused on metabolism. By the time the complete genome was sequenced (1997), only 26% of metabolic genes were accounted for in FBA models. Over the next 5 years the number grew to include nearly 80% of the metabolic genes. Methods for incorporating transcriptional regulation have been developed and implemented in a core metabolic model of E. coli, as have methods for including protein synthesis. Expanding the regulatory and protein synthesis models to the genome scale can be accomplished by using information that is known today (indicated by dotted lines). Further functional analysis of genes should increase the size of models (dashed lines). These three components can be combined to form an integrated model (E. coli i2K) that accounts for nearly 2,000 genes. The superscript letters indicate references, as follows: a, reference ; b, reference ; c, reference ; d, references and ; e, references and ; f, reference ; g, reference ; h, reference ; and i, reference .
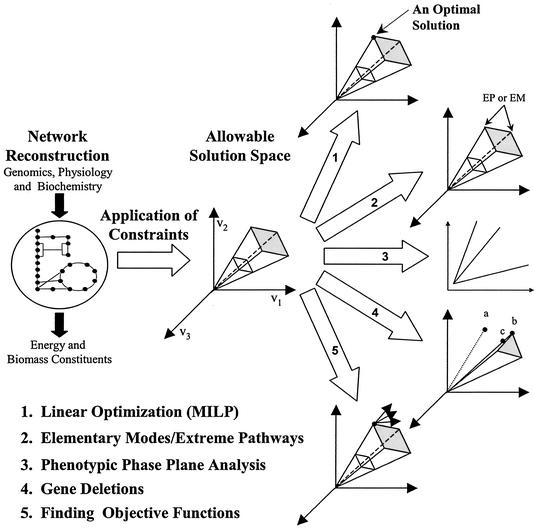
Constraint-based modeling. Application of constraints to a reconstructed metabolic network leads to a defined solution space in which a cell's network must operate. From this solution space a number of methods have been developed that help predict or explain phenotypic behavior. Linear optimization can be used to find solutions in the space that maximize or minimize a given objective (5, 14, 19, 62), and mixed-integer linear programming (MILP) can be used to find multiple optima if they exist (30, 41). Elementary mode analysis (52, 53) and extreme pathway analysis (50) can be used to characterize vectors in the solution space; the edges of the space correspond to extreme pathways (EP) and are a subset of the elementary modes (EM). Phenotypic phase plane analysis shows for what conditions the metabolic network operates under different limitations (18). The effects of gene deletions can also be computed. In the diagram the old optimal solution (point a) does not lie in the new solution space. A new optimum can be calculated (point b), or a suboptimal solution that is closest to the old optimum can be calculated (point c) (54). In addition, work has been done by using experimental flux measurements (indicated by a point) to back-calculate objective functions (indicated by vectors) (6a).
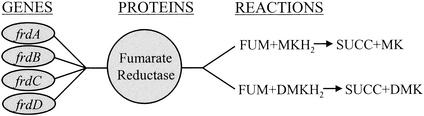
Gene-protein-reaction associations. The association between the enzyme fumarate reductase and the genes which code for its subunits is shown. All four gene products come together to make a functional enzyme. This enzyme is capable of carrying out two reactions, (i) the transfer of electrons from menaquinol (MKH2) to fumarate (FUM) and (ii) the transfer of electrons from demethylmenaquinol (DMKH2) to FUM. The products of both reactions are succinate (SUCC) and either menaquinone (MK) or demethylmenaquinone (DMK). Deletion of any of the subunits would eliminate the functional enzyme. This is simulated by removing the two reactions from the network (unless an isozyme exists).
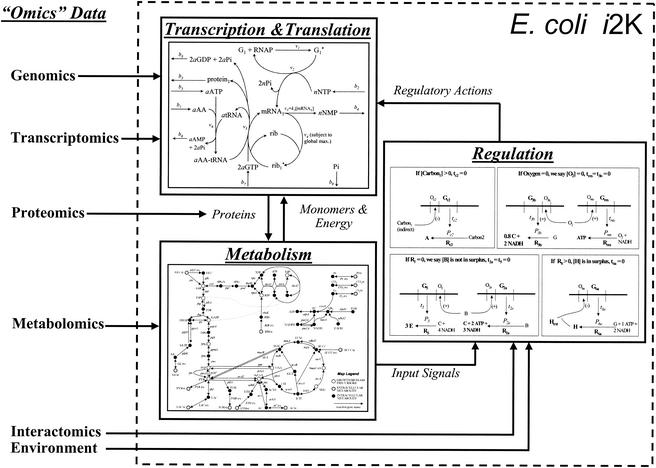
Integrated constraint-based model of E. coli: the E. coli i2K model. Constraint-based modeling frameworks have been developed for metabolism (5, 14, 19, 50, 52, 62), regulation (9), transcription, and translation (1). The connectivity among the three modeling components is shown here. Integration of these three modeling components should produce an integrated model of E. coli that accounts for nearly 2,000 genes, referred to as the E. coli i2K model. This model can be used to reconcile diverse “-omics” data and utilize the data to more accurately predict a cellular phenotype.
References
-
- Allen, T. E., and B. O. Palsson. 2003. Sequenced-based analysis of metabolic demands for protein synthesis in prokaryotes. J. Theor. Biol. 220:1-18. - PubMed
-
- Blattner, F. R., G. Plunkett 3rd, C. A. Bloch, N. T. Perna, V. Burland, M. Riley, J. Collado-Vides, J. D. Glasner, C. K. Rode, G. F. Mayhew, J. Gregor, N. W. Davis, H. A. Kirkpatrick, M. A. Goeden, D. J. Rose, B. Mau, and Y. Shao. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453-1474. - PubMed
-
- Bonarius, H. P. J., G. Schmid, and J. Tramper. 1997. Flux analysis of underdetermined metabolic networks: the quest for the missing constraints. Trends Biotechnol. 15:308-314.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
