

PTG gene deletion causes impaired glycogen synthesis and developmental insulin resistance
- PMID: 12727934
- PMCID: PMC154451
- DOI: 10.1172/JCI17975
PTG gene deletion causes impaired glycogen synthesis and developmental insulin resistance
Abstract
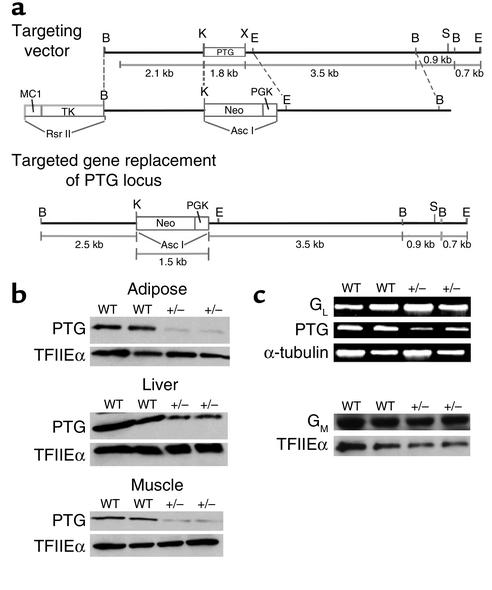
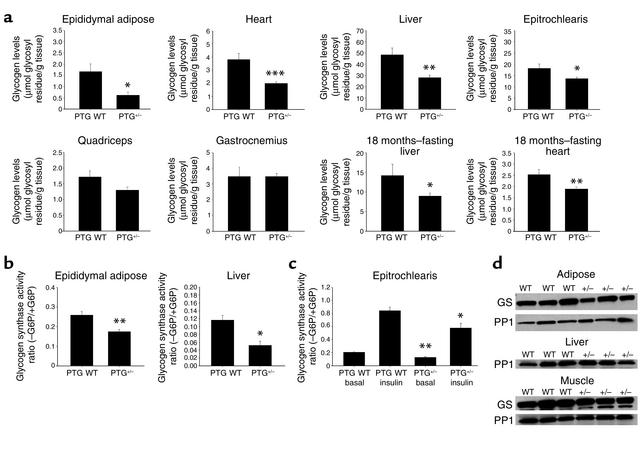
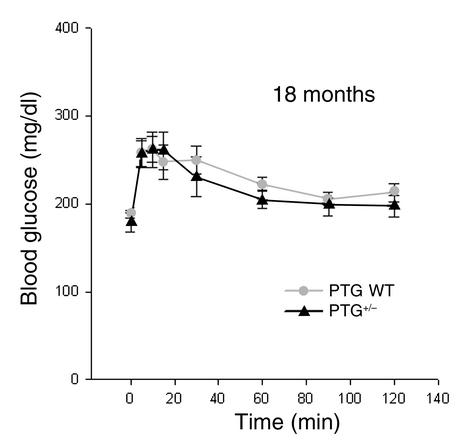
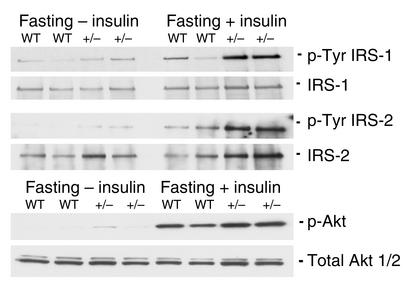
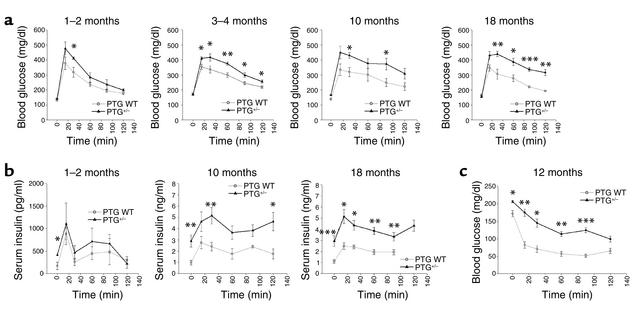
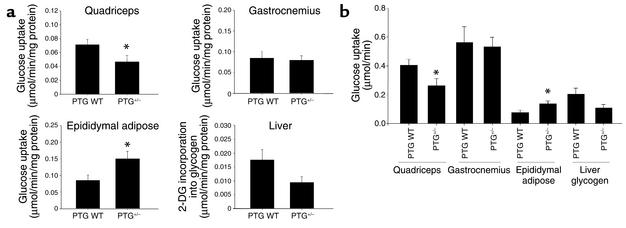
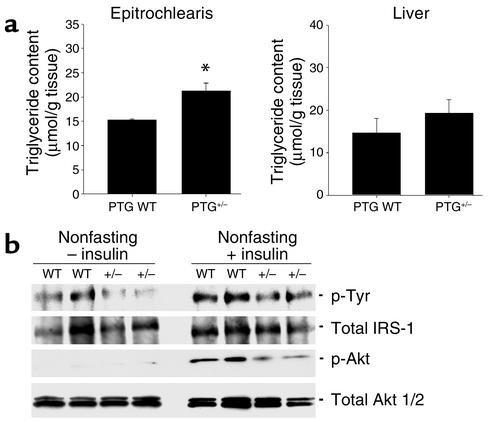
Protein targeting to glycogen (PTG) is a scaffolding protein that targets protein phosphatase 1alpha (PP1alpha) to glycogen, and links it to enzymes involved in glycogen synthesis and degradation. We generated mice that possess a heterozygous deletion of the PTG gene. These mice have reduced glycogen stores in adipose tissue, liver, heart, and skeletal muscle, corresponding with decreased glycogen synthase activity and glycogen synthesis rate. Although young PTG heterozygous mice initially demonstrate normal glucose tolerance, progressive glucose intolerance, hyperinsulinemia, and insulin resistance develop with aging. Insulin resistance in older PTG heterozygous mice correlates with a significant increase in muscle triglyceride content, with a corresponding attenuation of insulin receptor signaling. These data suggest that PTG plays a critical role in glycogen synthesis and is necessary to maintain the appropriate metabolic balance for the partitioning of fuel substrates between glycogen and lipid.
Figures
Comment in
-
Tissue glycogen content and glucose intolerance.J Clin Invest. 2003 May;111(9):1282-4. doi: 10.1172/JCI18526. J Clin Invest. 2003. PMID: 12727918 Free PMC article. No abstract available.
References
-
- Lawrence JC, Roach PJ. New insights into the role and mechanism of glycogen synthase activation by insulin. Diabetes. 1997;46:541–547. - PubMed
-
- Ingebritsen TS, Foulkes JG, Cohen P. The protein phosphatases involved in cellular regulation. 2. Glycogen metabolism. Eur. J. Biochem. 1983;132:263–274. - PubMed
-
- Krebs EG. Phosphorylation and dephosphorylation of glycogen phosphorylase: a prototype for reversible covalent enzyme modification. Curr. Top. Cell. Regul. 1981;18:401–419. - PubMed
-
- Dent P, et al. The molecular mechanism by which insulin stimulates glycogen synthesis in mammalian skeletal muscle. Nature. 1990;348:302–308. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
