

Heme oxygenase-2 protects against lipid peroxidation-mediated cell loss and impaired motor recovery after traumatic brain injury
- PMID: 12736340
- PMCID: PMC6742170
- DOI: 10.1523/JNEUROSCI.23-09-03689.2003
Heme oxygenase-2 protects against lipid peroxidation-mediated cell loss and impaired motor recovery after traumatic brain injury
Abstract
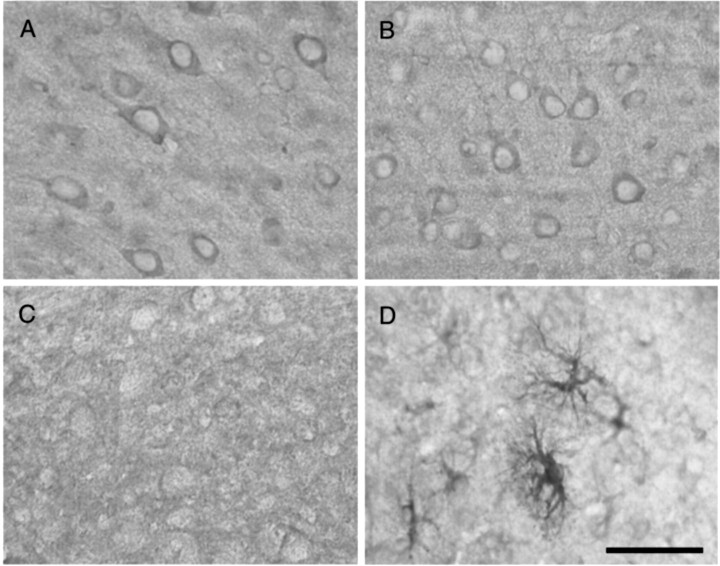
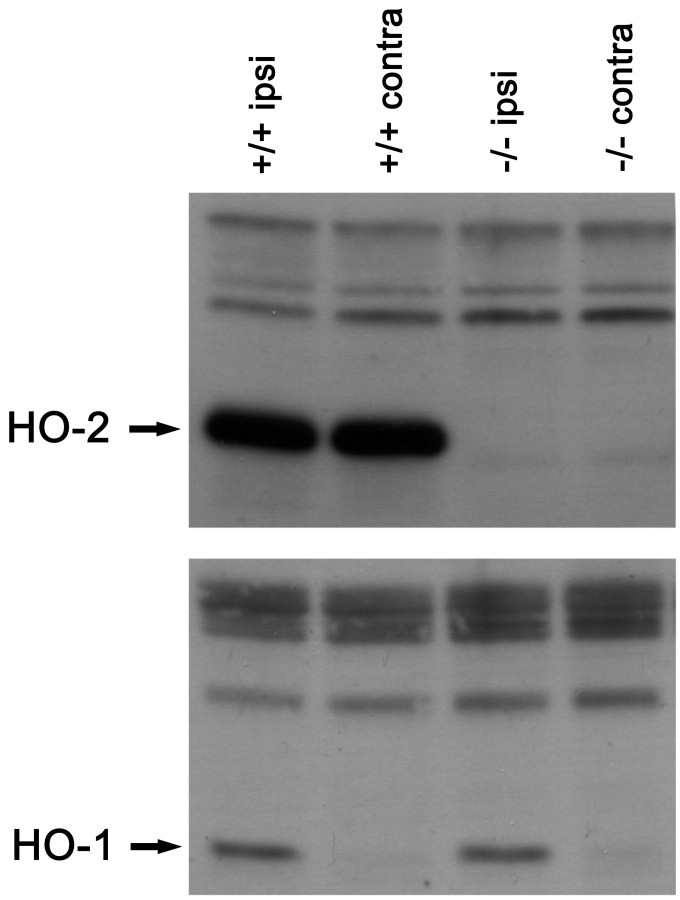
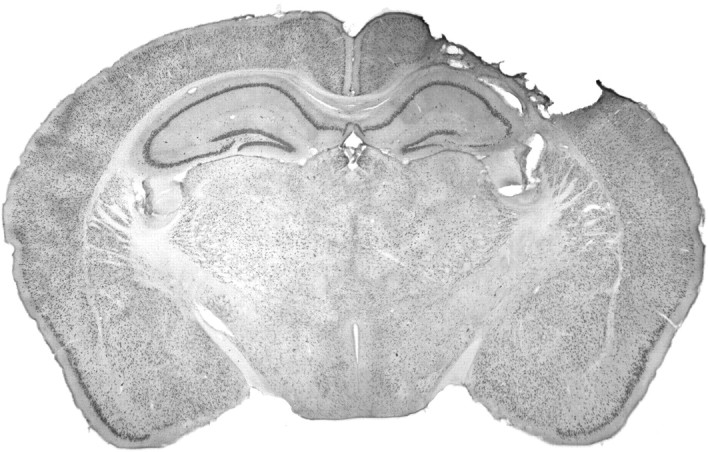
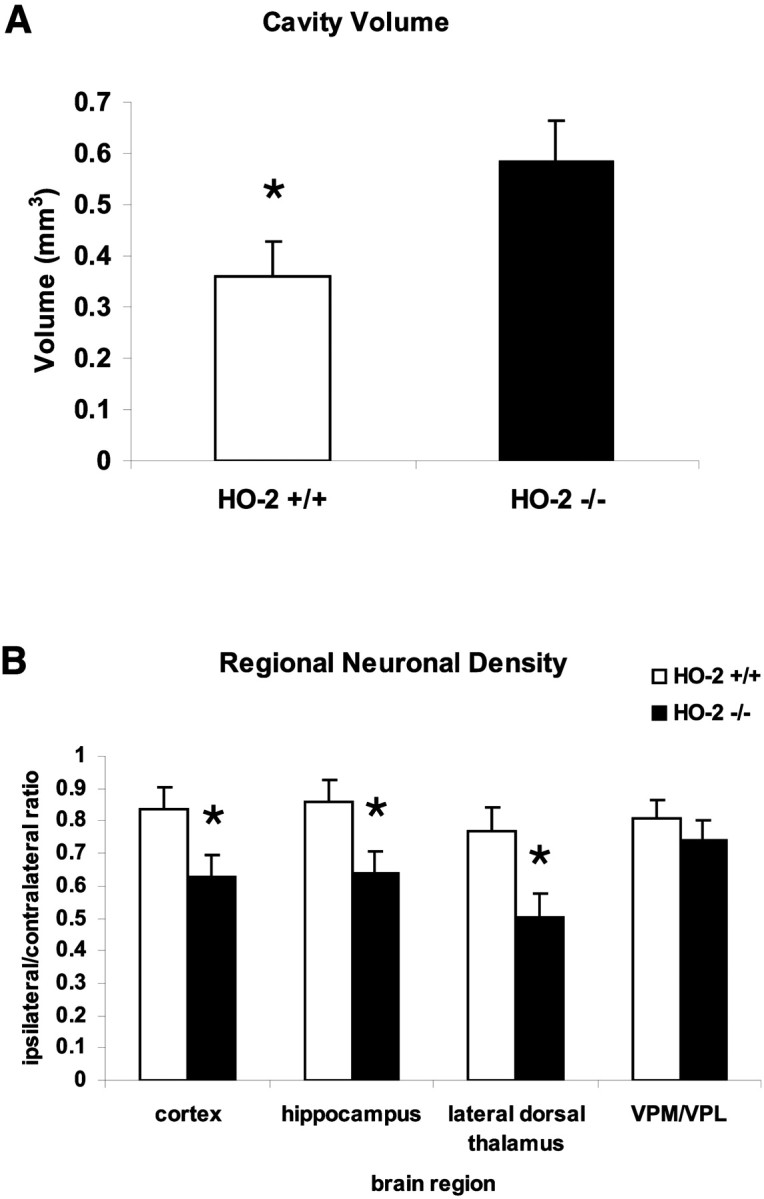
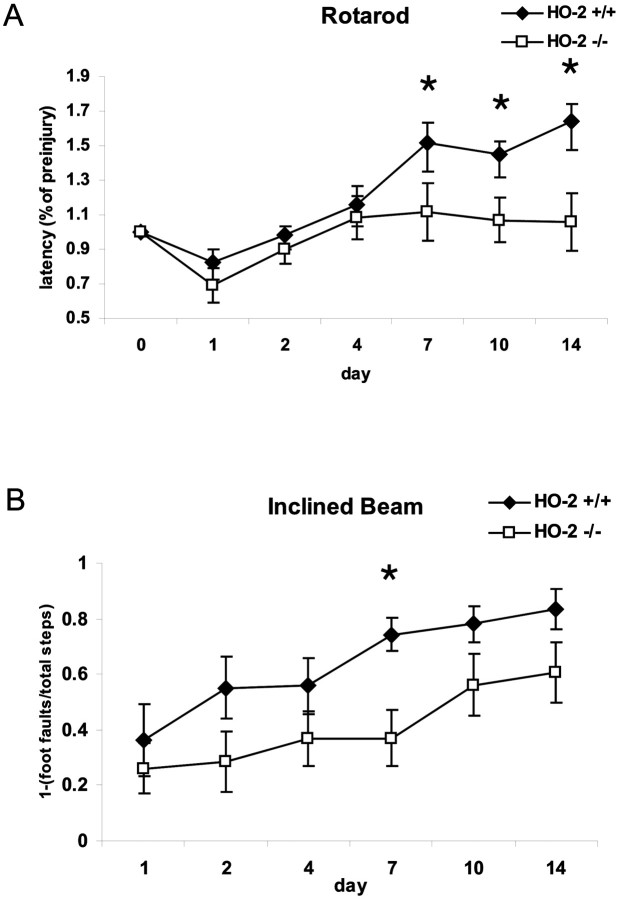
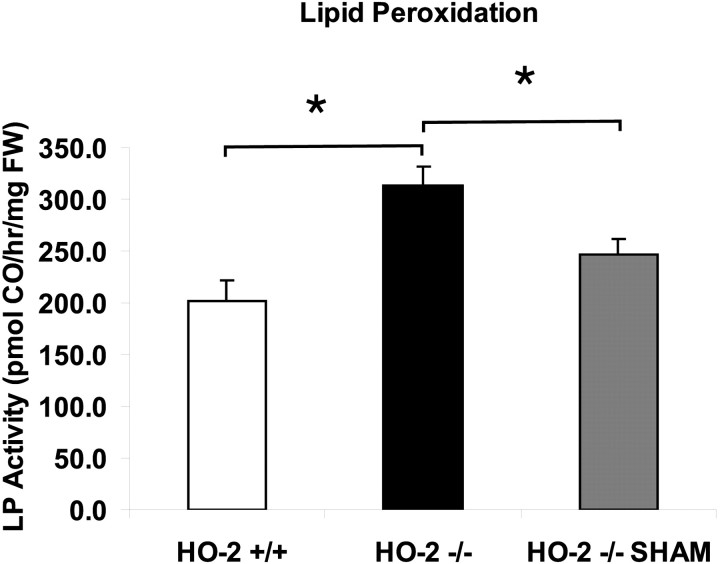
After traumatic brain injury (TBI), substantial extracellular heme is released from hemoproteins during hemorrhage and cell injury. Heme oxygenase (HO) isozymes are thought to detoxify the pro-oxidant heme to the potent antioxidant, bilirubin. HO-1, the inducible isozyme, is expressed in glial populations after injury and may play a protective role. However, the role of HO-2, the predominant and constitutively expressed isozyme in the brain, remains unclear after TBI. We used a controlled cortical impact injury model to determine the extent and mechanism of damage between HO-2 knock-out (KO) (-/-) and wild-type (WT) (+/+) mice. The specific cellular and temporal expressions of HO-2 and HO-1 were characterized by immunocytochemistry and Western blots. HO-2 was immunolocalized in neurons both before and after TBI, whereas HO-1 was highly upregulated in glia only after TBI. HO activity determined by gas chromatography using brain sonicates from injured HO-2 KO mice was significantly less than that of HO-2 wild types, despite the induction of HO-1 expression after TBI. Cell loss was significantly greater in KO mice in areas including the cortex, the CA3 region of hippocampus, and the lateral dorsal thalamus. Furthermore, motor recovery after injury, as measured by the rotarod assay and an inclined beam-walking task, was compromised in the KO mice. Finally, brain tissue from injured HO-2 KO mice exhibited decreased ability to reduce oxidative stress, as measured with an Fe(2+)/ascorbic acid-mediated carbon monoxide generation assay for lipid peroxidation susceptibility. These findings demonstrate that HO-2 expression protects neurons against TBI by reducing lipid peroxidation via the catabolism of free heme.
Figures

References
-
- Burnett AL, Johns DG, Kriegsfeld LJ, Klein SL, Calvin DC, Demas GE, Schramm LP, Tonegawa S, Nelson RJ, Snyder SH, Poss KD. Ejaculatory abnormalities in mice with targeted disruption of the gene for heme oxygenase-2. Nat Med. 1998;4:84–87. - PubMed
-
- Dennery PA. Regulation and role of heme oxygenase in oxidative injury. Curr Top Cell Regul. 2000;36:181–199. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous