

Activation of Mst1 causes dilated cardiomyopathy by stimulating apoptosis without compensatory ventricular myocyte hypertrophy
- PMID: 12750396
- PMCID: PMC155047
- DOI: 10.1172/JCI17459
Activation of Mst1 causes dilated cardiomyopathy by stimulating apoptosis without compensatory ventricular myocyte hypertrophy
Abstract
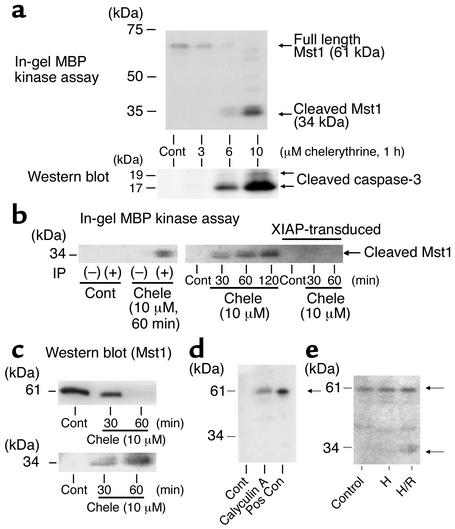
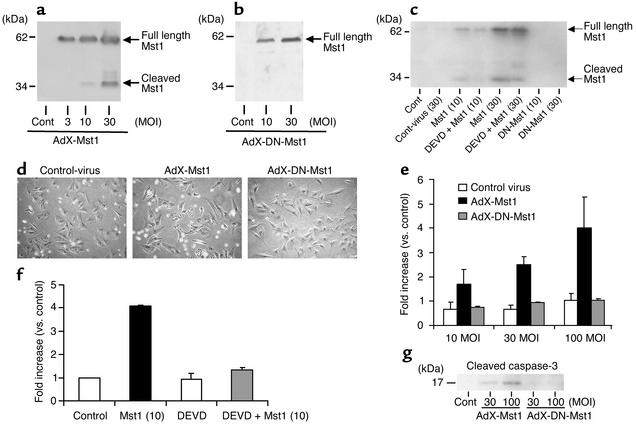
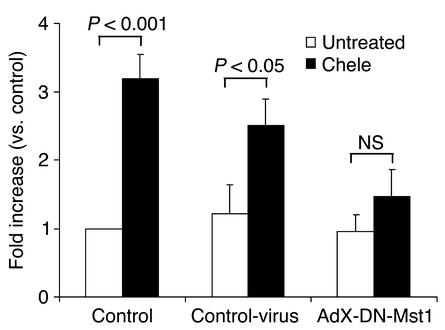
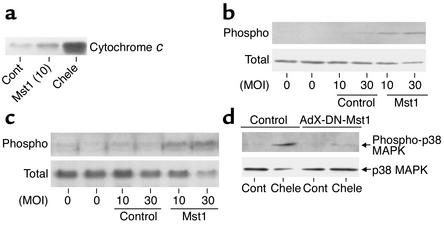
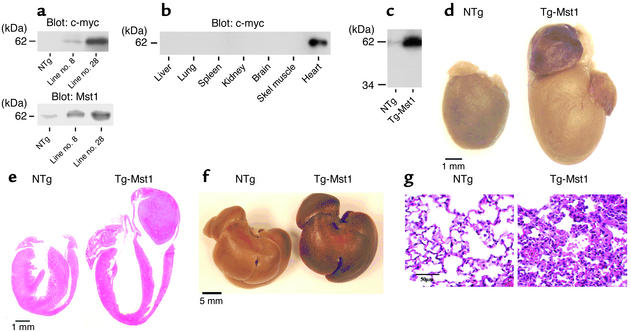
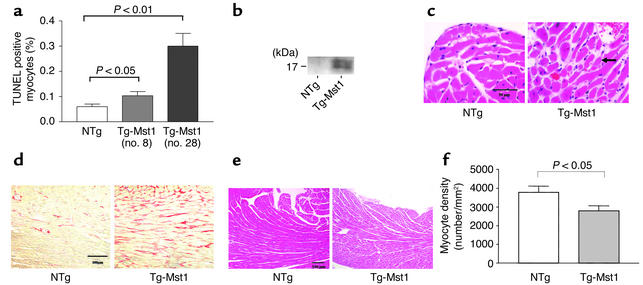
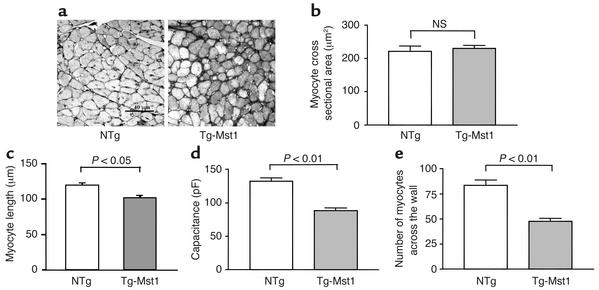
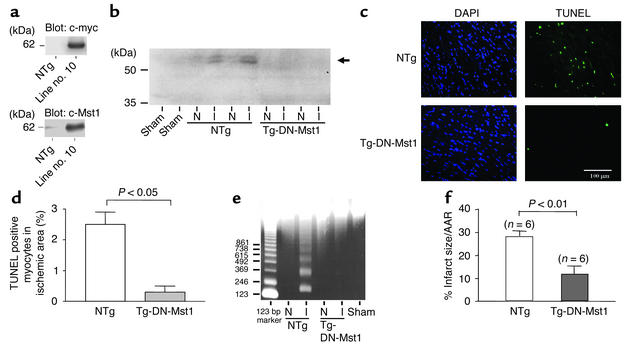
Activation of mammalian sterile 20-like kinase 1 (Mst1) by genotoxic compounds is known to stimulate apoptosis in some cell types. The importance of Mst1 in cell death caused by clinically relevant pathologic stimuli is unknown, however. In this study, we show that Mst1 is a prominent myelin basic protein kinase activated by proapoptotic stimuli in cardiac myocytes and that Mst1 causes cardiac myocyte apoptosis in vitro in a kinase activity-dependent manner. In vivo, cardiac-specific overexpression of Mst1 in transgenic mice results in activation of caspases, increased apoptosis, and dilated cardiomyopathy. Surprisingly, however, Mst1 prevents compensatory cardiac myocyte elongation or hypertrophy despite increased wall stress, thereby obscuring the use of the Frank-Starling mechanism, a fundamental mechanism by which the heart maintains cardiac output in response to increased mechanical load at the single myocyte level. Furthermore, Mst1 is activated by ischemia/reperfusion in the mouse heart in vivo. Suppression of endogenous Mst1 by cardiac-specific overexpression of dominant-negative Mst1 in transgenic mice prevents myocyte death by pathologic insults. These results show that Mst1 works as both an essential initiator of apoptosis and an inhibitor of hypertrophy in cardiac myocytes, resulting in a previously unrecognized form of cardiomyopathy.
Figures
Comment in
-
A matter of life and death: cardiac myocyte apoptosis and regeneration.J Clin Invest. 2003 May;111(10):1457-9. doi: 10.1172/JCI18611. J Clin Invest. 2003. PMID: 12750394 Free PMC article. No abstract available.
References
-
- Kajstura J, et al. Apoptotic and necrotic myocyte cell death are independent contributing variables of infarct size in rats. Lab. Invest. 1996;74:86–107. - PubMed
-
- Saraste A, et al. Apoptosis in human acute myocardial infarction. Circulation. 1997;95:320–323. - PubMed
-
- Tanaka M, et al. Hypoxia induces apoptosis with enhanced expression of fas antigen messenger RNA in cultured neonatal rat cardiomyocytes. Circ. Res. 1994;75:426–433. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R37 HL033107/HL/NHLBI NIH HHS/United States
- HL-67727/HL/NHLBI NIH HHS/United States
- HL-67724/HL/NHLBI NIH HHS/United States
- HL-69020/HL/NHLBI NIH HHS/United States
- R01 HL033107/HL/NHLBI NIH HHS/United States
- AG-14121/AG/NIA NIH HHS/United States
- HL-33107/HL/NHLBI NIH HHS/United States
- HL-59139/HL/NHLBI NIH HHS/United States
- HL-65183/HL/NHLBI NIH HHS/United States
- P01 HL069020/HL/NHLBI NIH HHS/United States
- HL-33065/HL/NHLBI NIH HHS/United States
- R01 HL067724/HL/NHLBI NIH HHS/United States
- R01 HL065182/HL/NHLBI NIH HHS/United States
- P01 HL059139/HL/NHLBI NIH HHS/United States
- HL-65182/HL/NHLBI NIH HHS/United States
- R01 AG014121/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
