

Modeling bacterial evolution with comparative-genome-based marker systems: application to Mycobacterium tuberculosis evolution and pathogenesis
- PMID: 12754238
- PMCID: PMC155390
- DOI: 10.1128/JB.185.11.3392-3399.2003
Modeling bacterial evolution with comparative-genome-based marker systems: application to Mycobacterium tuberculosis evolution and pathogenesis
Abstract
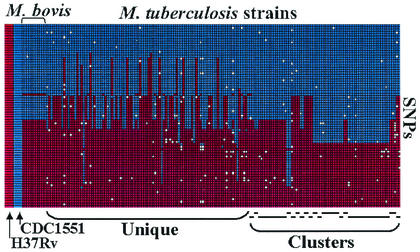
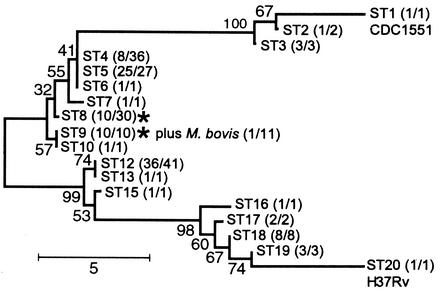
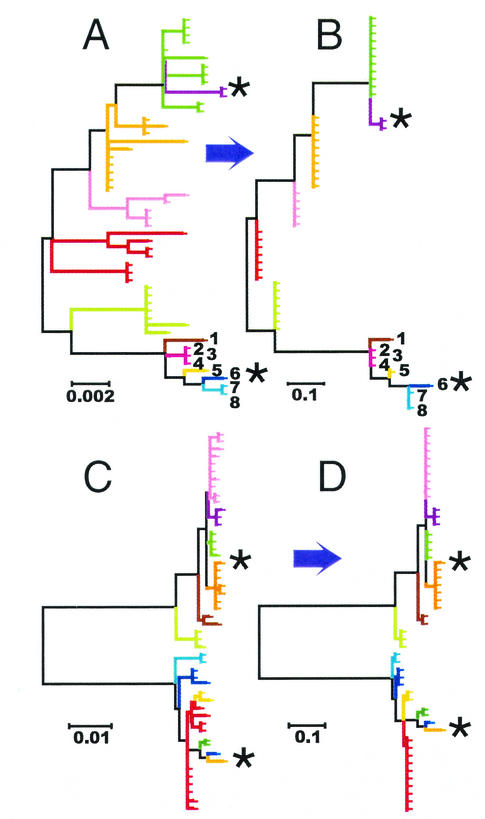
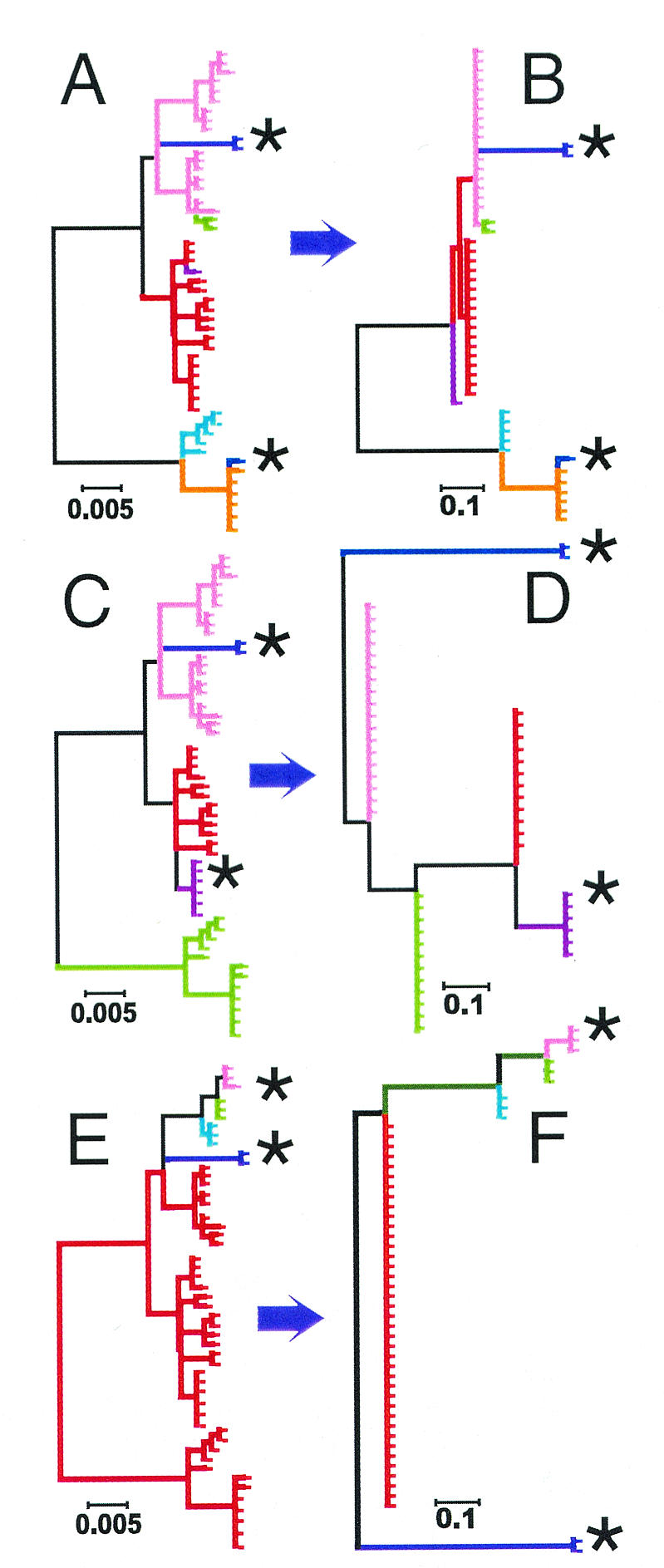
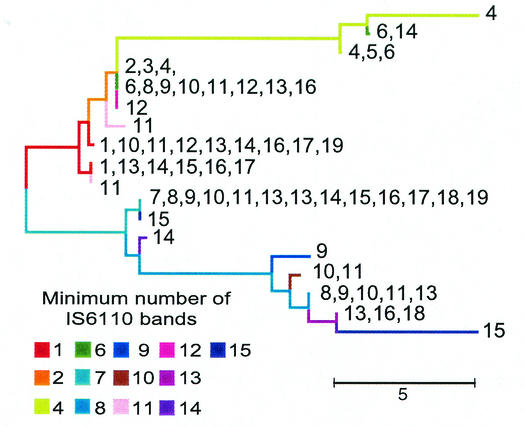
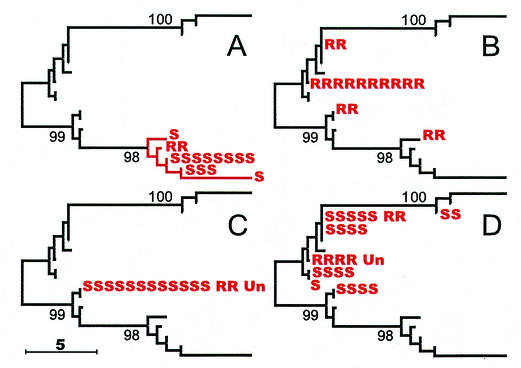
The comparative-genomic sequencing of two Mycobacterium tuberculosis strains enabled us to identify single nucleotide polymorphism (SNP) markers for studies of evolution, pathogenesis, and epidemiology in clinical M. tuberculosis. Phylogenetic analysis using these "comparative-genome markers" (CGMs) produced a highly unusual phylogeny with a complete absence of secondary branches. To investigate CGM-based phylogenies, we devised computer models to simulate sequence evolution and calculate new phylogenies based on an SNP format. We found that CGMs represent a distinct class of phylogenetic markers that depend critically on the genetic distances between compared "reference strains." Properly distanced reference strains generate CGMs that accurately depict evolutionary relationships, distorted only by branch collapse. Improperly distanced reference strains generate CGMs that distort and reroot outgroups. Applying this understanding to the CGM-based phylogeny of M. tuberculosis, we found evidence to suggest that this species is highly clonal without detectable lateral gene exchange. We noted indications of evolutionary bottlenecks, including one at the level of the PHRI "C" strain previously associated with particular virulence characteristics. Our evidence also suggests that loss of IS6110 to fewer than seven elements per genome is uncommon. Finally, we present population-based evidence that KasA, an important component of mycolic acid biosynthesis, develops G312S polymorphisms under selective pressure.
Figures
References
-
- Alland, D., G. E. Kalkut, A. R. Moss, R. A. McAdam, J. A. Hahn, W. Bosworth, E. Drucker, and B. R. Bloom. 1994. Transmission of tuberculosis in New York City. An analysis by DNA fingerprinting and conventional epidemiologic methods. N. Engl. J. Med. 330:1710-1716. - PubMed
-
- Brosch, R., S. V. Gordon, M. Marmiesse, P. Brodin, C. Buchrieser, K. Eiglmeier, T. Garnier, C. Gutierrez, G. G. Hewinson, K. Kremer, L. M. Parsons, A. S. Pym, S. Samper, D. van Soolingen, and S. T. Cole. 2002. A new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc. Natl. Acad. Sci. USA 99:3684-3689. - PMC - PubMed
-
- Cole, S. T., R. Brosch, J. Parkhill, T. Garnier, C. Churcher, D. Harris, S. V. Gordon, L. Eiglmeier, S. Gas, C. E. Barry III, F. Tekaia, K. Badcock, D. Basham, D. Brown, T. Chillingworth, R. Connor, R. Davies, K. Devlin, T. Feltwell, S. Gentles, N. Hamlin, S. Holroyd, T. Hornsby, K. Jagels, A. Krogh, J. Mclean, S. Moule, L. Murphy, K. Oliver, J. Osborne, M. A. Quai, M. A. Rajandream, J. Rogers, S. Rutter, K. Seeger, J. Skelton, R. Squares, S. Squares, J. E. Sulston, K. Taylor, S. Whitehead, and B. G. Barrell. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393:537-544. - PubMed
-
- Cummings, C. A., and D. A. Relman. 2002. Genomics and microbiology. Microbial forensics—“cross-examining pathogens.” Science 296:1976-1979. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
