

NKG2D-mediated natural killer cell protection against cytomegalovirus is impaired by viral gp40 modulation of retinoic acid early inducible 1 gene molecules
- PMID: 12756263
- PMCID: PMC2193789
- DOI: 10.1084/jem.20021973
NKG2D-mediated natural killer cell protection against cytomegalovirus is impaired by viral gp40 modulation of retinoic acid early inducible 1 gene molecules
Abstract
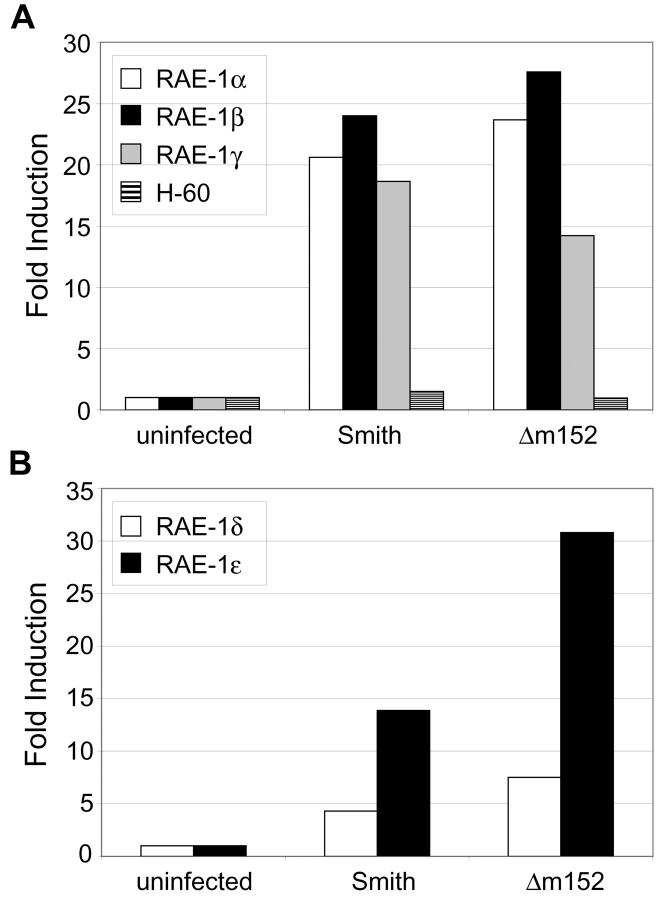
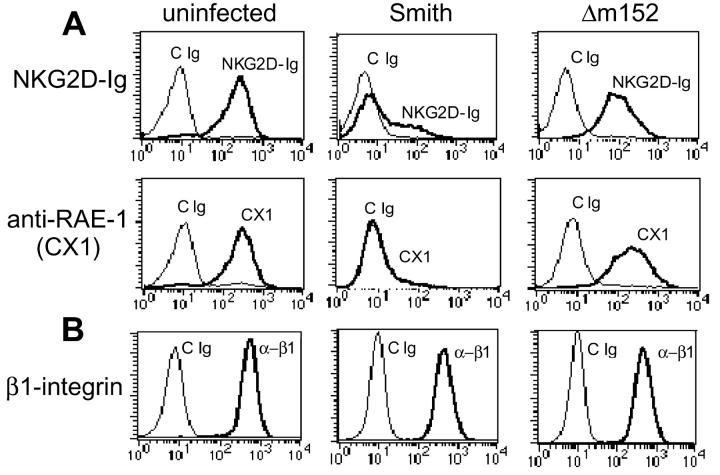
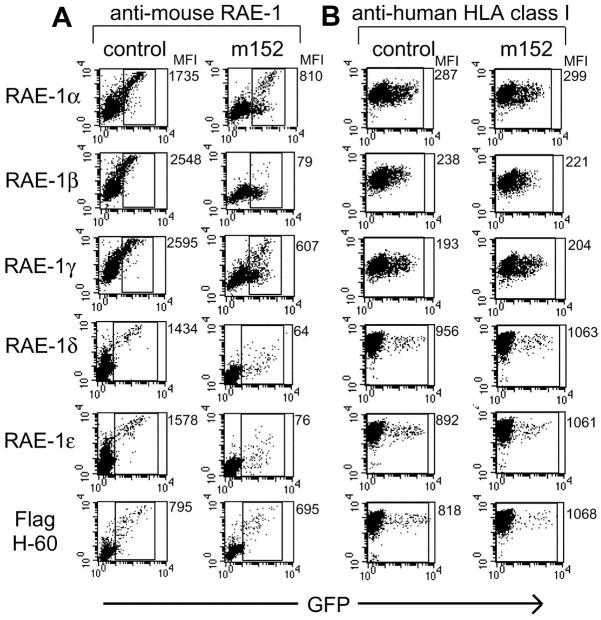
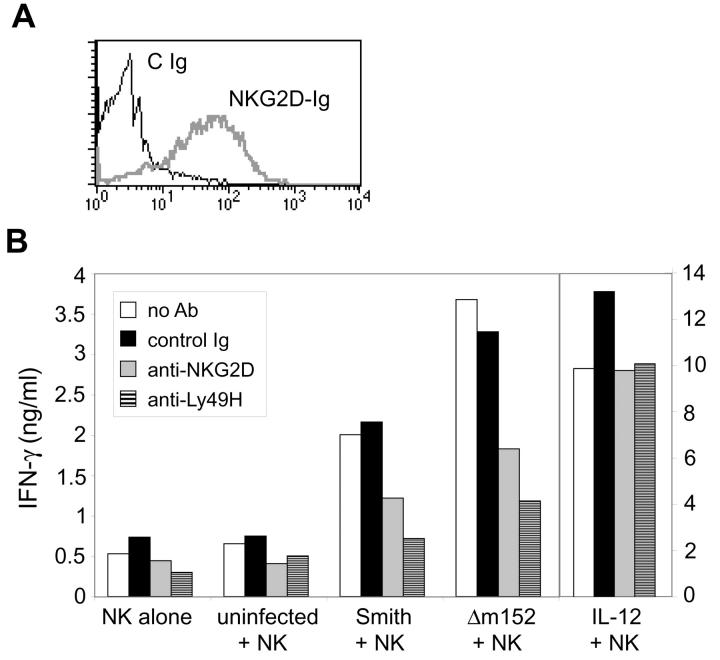
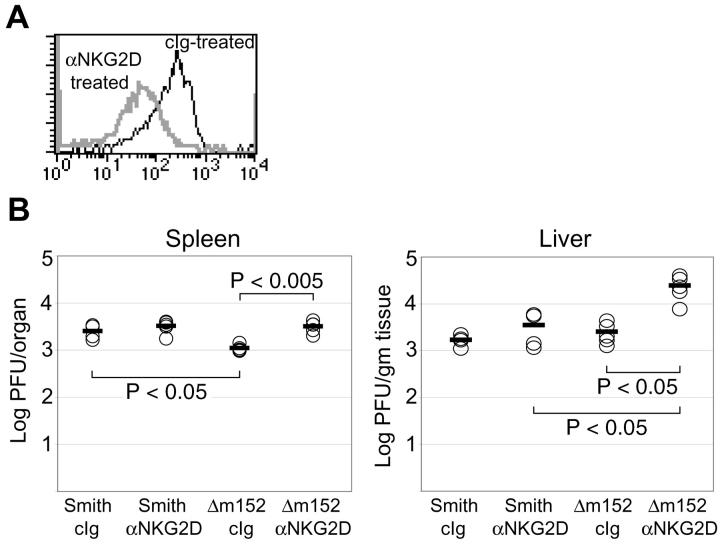
Natural killer (NK) cells play a critical role in the innate immune response against cytomegalovirus (CMV) infections. Although CMV encodes several gene products committed to evasion of adaptive immunity, viral modulation of NK cell activity is only beginning to be appreciated. A previous study demonstrated that the mouse CMV m152-encoded gp40 glycoprotein diminished expression of ligands for the activating NK cell receptor NKG2D on the surface of virus-infected cells. Here we have defined the precise ligands that are affected and have directly implicated NKG2D in immune responses to CMV infection in vitro and in vivo. Murine CMV (MCMV) infection potently induced transcription of all five known retinoic acid early inducible 1 (RAE-1) genes (RAE-1alpha, RAE-1beta, RAE-1delta, RAE-1 epsilon, and RAE-1gamma), but not H-60. gp40 specifically down-regulated the cell surface expression of all RAE-1 proteins, but not H-60, and diminished NK cell interferon gamma production against CMV-infected cells. Consistent with previous findings, a m152 deletion mutant virus (Deltam152) was less virulent in vivo than the wild-type Smith strain of MCMV. Treatment of BALB/c mice with a neutralizing anti-NKG2D antibody before infection increased titers of Deltam152 virus in the spleen and liver to levels seen with wild-type virus. These experiments demonstrate that gp40 impairs NK cell recognition of virus-infected cells through disrupting the RAE-1-NKG2D interaction.
Figures
References
-
- Tortorella, D., B.E. Gewurz, M.H. Furman, D.J. Schust, and H.L. Ploegh. 2000. Viral subversion of the immune system. Annu. Rev. Immunol. 18:861–926. - PubMed
-
- Mocarski, E.S., Jr. 2002. Immunomodulation by cytomegaloviruses: manipulative strategies beyond evasion. Trends Microbiol. 10:332–339. - PubMed
-
- Kleijnen, M.F., J.B. Huppa, P. Lucin, S. Mukherjee, H. Farrell, A.E. Campbell, U.H. Koszinowski, A.B. Hill, and H.L. Ploegh. 1997. A mouse cytomegalovirus glycoprotein, gp34, forms a complex with folded class I MHC molecules in the ER which is not retained but is transported to the cell surface. EMBO J. 16:685–694. - PMC - PubMed
-
- Kavanagh, D.G., U.H. Koszinowski, and A.B. Hill. 2001. The murine cytomegalovirus immune evasion protein m4/gp34 forms biochemically distinct complexes with class I MHC at the cell surface and in a pre-Golgi compartment. J. Immunol. 167:3894–3902. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
