

Mutational analysis of the enzymatic domain of Clostridium difficile toxin B reveals novel inhibitors of the wild-type toxin
- PMID: 12761111
- PMCID: PMC155706
- DOI: 10.1128/IAI.71.6.3294-3301.2003
Mutational analysis of the enzymatic domain of Clostridium difficile toxin B reveals novel inhibitors of the wild-type toxin
Abstract
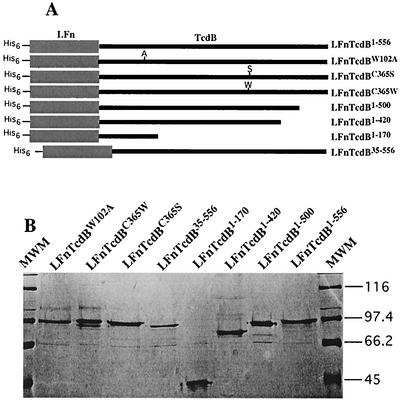
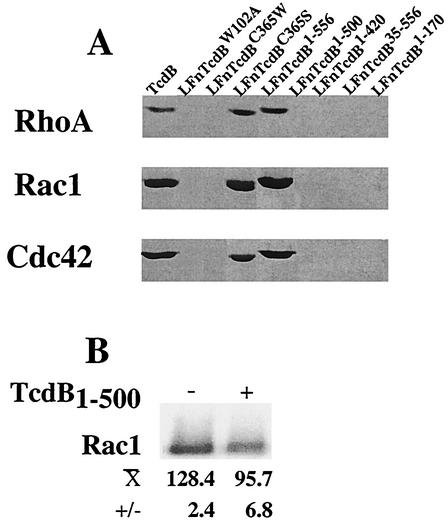
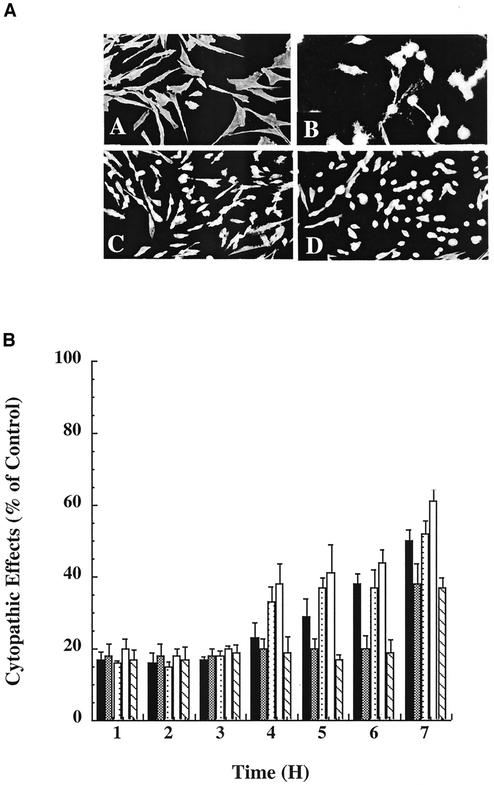
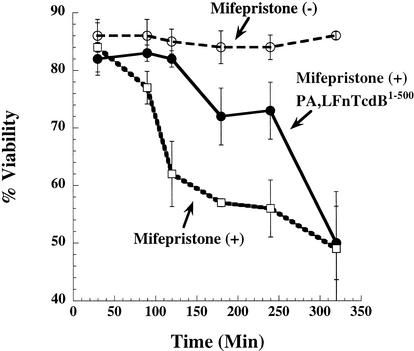
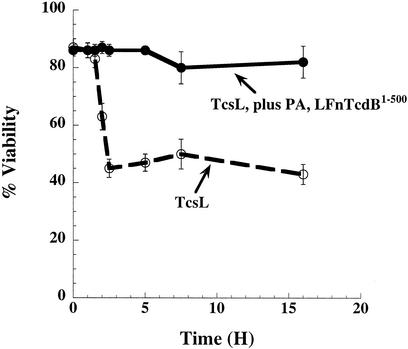
Toxin B (TcdB), a major Clostridium difficile virulence factor, glucosylates and inactivates the small GTP-binding proteins Rho, Rac, and Cdc42. In the present study we provide evidence that enzymatically inactive fragments of the TcdB enzymatic domain are effective intracellular inhibitors of native TcdB. Site-directed and deletion mutants of the TcdB enzymatic region (residues 1 to 556), lacking receptor binding and cell entry domains, were analyzed for attenuation of glucosyltransferase and glucosylhydrolase activity. Five of six derivatives from TcdB(1-556) were found to be devoid of enzymatic activity. In order to facilitate cell entry, mutants were genetically fused to lfn, which encodes the protective antigen binding region of anthrax toxin lethal factor and mediates the cell entry of heterologous proteins. In line with reduced enzymatic activity, the mutants also lacked cytotoxicity. Remarkably, pretreatment or cotreatment of cells with four of the mutants provided protection against the cytotoxic effects of native TcdB. Furthermore, a CHO cell line expressing enzymatically active TcdB(1-556) was also protected by the mutant-derived inhibitors, suggesting that inhibition occurred at an intracellular location. Protection also was afforded by the inhibitor to cells treated with Clostridium sordellii lethal toxin (TcsL), which uses the same cosubstrate as TcdB but shares Rac only as a common substrate target. Finally, the inhibitor did not provide protection against Clostridium novyi alpha-toxin (Tcnalpha), which shares similar substrates with TcdB yet uses a different cosubstrate. This is the first report to demonstrate that the potential exists to inhibit toxins at their intracellular site of action by using inactive mutants.
Figures

Similar articles
-
Cytosolic delivery and characterization of the TcdB glucosylating domain by using a heterologous protein fusion.Infect Immun. 2001 Jan;69(1):599-601. doi: 10.1128/IAI.69.1.599-601.2001. Infect Immun. 2001. PMID: 11119561 Free PMC article.
-
Specific inhibition of phorbol ester-stimulated phospholipase D by Clostridium sordellii lethal toxin and Clostridium difficile toxin B-1470 in HEK-293 cells. Restoration by Ral GTPases.J Biol Chem. 1998 Mar 27;273(13):7413-22. doi: 10.1074/jbc.273.13.7413. J Biol Chem. 1998. PMID: 9516439
-
Haemorrhagic toxin and lethal toxin from Clostridium sordellii strain vpi9048: molecular characterization and comparative analysis of substrate specificity of the large clostridial glucosylating toxins.Cell Microbiol. 2014 Nov;16(11):1706-21. doi: 10.1111/cmi.12321. Epub 2014 Aug 4. Cell Microbiol. 2014. PMID: 24905543
-
The Role of Rho GTPases in Toxicity of Clostridium difficile Toxins.Toxins (Basel). 2015 Dec 2;7(12):5254-67. doi: 10.3390/toxins7124874. Toxins (Basel). 2015. PMID: 26633511 Free PMC article. Review.
-
Clostridium difficile Toxin Biology.Annu Rev Microbiol. 2017 Sep 8;71:281-307. doi: 10.1146/annurev-micro-090816-093458. Epub 2017 Jun 28. Annu Rev Microbiol. 2017. PMID: 28657883 Review.
Cited by
-
Dominant-negative inhibitors of the Clostridium perfringens epsilon-toxin.J Biol Chem. 2009 Oct 23;284(43):29446-53. doi: 10.1074/jbc.M109.021782. Epub 2009 Aug 31. J Biol Chem. 2009. PMID: 19720828 Free PMC article.
-
Clostridium difficile toxin glucosyltransferase domains in complex with a non-hydrolyzable UDP-glucose analogue.J Struct Biol. 2017 Jun;198(3):203-209. doi: 10.1016/j.jsb.2017.04.006. Epub 2017 Apr 19. J Struct Biol. 2017. PMID: 28433497 Free PMC article.
-
Clostridium difficile toxins: mechanism of action and role in disease.Clin Microbiol Rev. 2005 Apr;18(2):247-63. doi: 10.1128/CMR.18.2.247-263.2005. Clin Microbiol Rev. 2005. PMID: 15831824 Free PMC article. Review.
-
Development of a Novel Vaccine Containing Binary Toxin for the Prevention of Clostridium difficile Disease with Enhanced Efficacy against NAP1 Strains.PLoS One. 2017 Jan 26;12(1):e0170640. doi: 10.1371/journal.pone.0170640. eCollection 2017. PLoS One. 2017. PMID: 28125650 Free PMC article.
-
Glucosyltransferase Activity of Clostridium difficile Toxin B Triggers Autophagy-mediated Cell Growth Arrest.Sci Rep. 2017 Sep 5;7(1):10532. doi: 10.1038/s41598-017-11336-4. Sci Rep. 2017. PMID: 28874882 Free PMC article.
References
-
- Arora, N., K. R. Klimpel, Y. Singh, and S. H. Leppla. 1992. Fusions of anthrax toxin lethal factor to the ADP-ribosylation domain of Pseudomonas exotoxin A are potent cytotoxins which are translocated to the cytosol of mammalian cells. J. Biol. Chem. 267:15542-15548. - PubMed
-
- Arora, N., and S. H. Leppla. 1993. Residues 1-254 of anthrax toxin lethal factor are sufficient to cause cellular uptake of fused polypeptides. J. Biol. Chem. 268:3334-3341. - PubMed
-
- Arora, N., L. C. Williamson, S. H. Leppla, and J. L. Halpern. 1994. Cytotoxic effects of a chimeric protein consisting of tetanus toxin light chain and anthrax toxin lethal factor in non-neuronal cells. J. Biol. Chem. 269:26165-26171. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
