

Automated protein fold determination using a minimal NMR constraint strategy
- PMID: 12761394
- PMCID: PMC2323888
- DOI: 10.1110/ps.0300203
Automated protein fold determination using a minimal NMR constraint strategy
Abstract
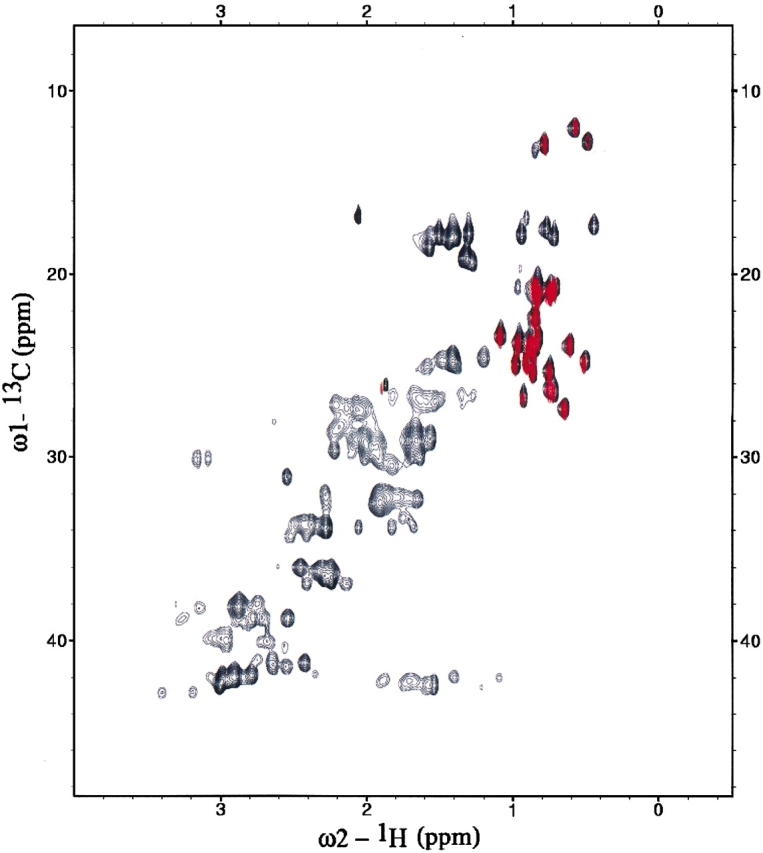
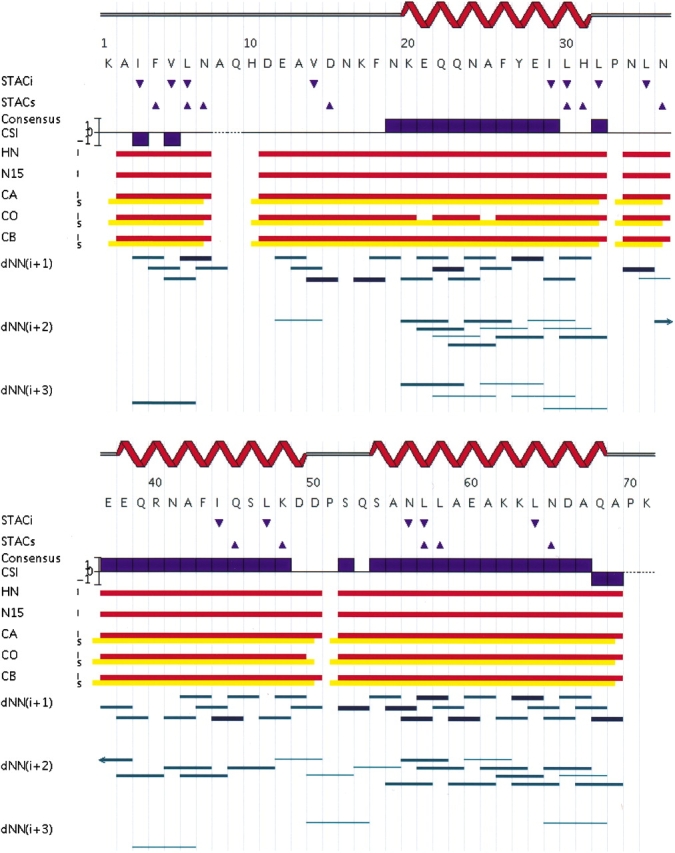
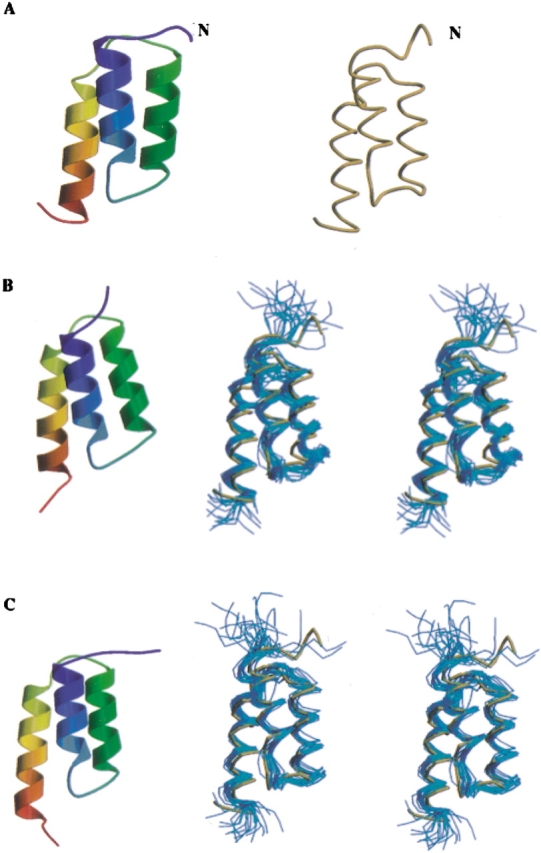
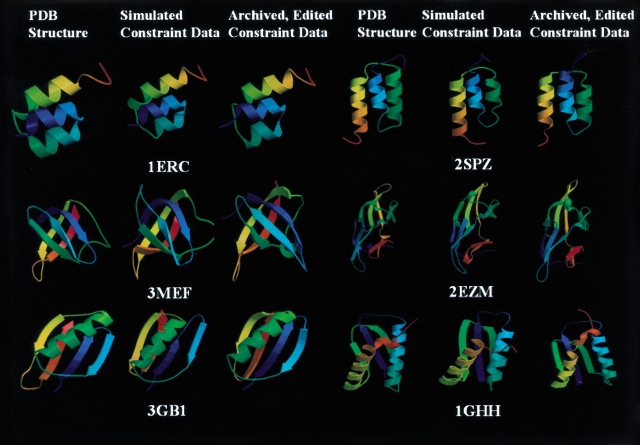
Determination of precise and accurate protein structures by NMR generally requires weeks or even months to acquire and interpret all the necessary NMR data. However, even medium-accuracy fold information can often provide key clues about protein evolution and biochemical function(s). In this article we describe a largely automatic strategy for rapid determination of medium-accuracy protein backbone structures. Our strategy derives from ideas originally introduced by other groups for determining medium-accuracy NMR structures of large proteins using deuterated, (13)C-, (15)N-enriched protein samples with selective protonation of side-chain methyl groups ((13)CH(3)). Data collection includes acquiring NMR spectra for automatically determining assignments of backbone and side-chain (15)N, H(N) resonances, and side-chain (13)CH(3) methyl resonances. These assignments are determined automatically by the program AutoAssign using backbone triple resonance NMR data, together with Spin System Type Assignment Constraints (STACs) derived from side-chain triple-resonance experiments. The program AutoStructure then derives conformational constraints using these chemical shifts, amide (1)H/(2)H exchange, nuclear Overhauser effect spectroscopy (NOESY), and residual dipolar coupling data. The total time required for collecting such NMR data can potentially be as short as a few days. Here we demonstrate an integrated set of NMR software which can process these NMR spectra, carry out resonance assignments, interpret NOESY data, and generate medium-accuracy structures within a few days. The feasibility of this combined data collection and analysis strategy starting from raw NMR time domain data was illustrated by automatic analysis of a medium accuracy structure of the Z domain of Staphylococcal protein A.
Figures
References
-
- Aghazadeh, B., Zhu, K., Kubiseski, T.J., Liu, G.A., Pawson, T., Zheng, Y., and Rosen, M.K. 1998. Structure and mutagenesis of the DbI homology domain. Nat. Struct. Biol. 5 1098–1107. - PubMed
-
- Andrec, M., Du, P., and Levy, R.M. 2001. Protein backbone structure determination using only residual dipolar couplings from one ordering medium. J. Biomol. NMR 21 335–347. - PubMed
-
- Andrec, M., Harano, Y., Jacobson, M.P., Friesner, R., and Levy, R.M. 2002. Complete protein structure determination using backbone residual dipolar couplings and sidechain rotamer prediction. J. Struct. Funct. Genomics 2 103–111. - PubMed
-
- Berardi, M.J., Sun, C., Zehr, M., Abildgaard, F., Peng, J., Speck, N.A., and Bushweller, J.H. 1999. The Ig fold of the core binding factor α Runt domain is a member of a family of structurally and functionally related Ig-fold DNA-binding domains. Structure 7 1247–1256. - PubMed
-
- Brunger, A.T., Adams, P.D., Clore, G.M., Delano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta. Crystallogr. D54 905–921. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
